1000/1000
Hot
Most Recent
 +1 point
+1 point
Merkel cell polyomavirus (MCV) is the only known human oncogenic virus in the polyomaviridae family and the etiological agent of most Merkel cell carcinomas (MCC). MCC is an aggressive and highly metastatic skin cancer with a propensity for recurrence and poor prognosis. Large tumor antigen (LT), is an essential oncoprotein for MCV transcription, viral replication, and cancer cell proliferation. MCV LT is a short-lived protein that encodes a unique domain: MCV LT unique regions (MURs). These domains consist of phosphorylation sites that interact with multiple E3 ligases, thus limiting LT expression and consequently, viral replication. In this study, we show that MURs are necessary for regulating LT stability via multiple E3 ligase interactions, resulting in cell growth arrest. While expression of wild-type MCV LT induced a decrease in cellular proliferation, deletion of the MUR domains resulted in increased LT stability and cell proliferation. Conversely, addition of MURs to SV40 LT propagated E3 ligase interactions, which in turn, reduced SV40 LT stability and decreased cell growth activity. Our results demonstrate that compared to other human polyomaviruses (HPyVs), MCV LT has evolved to acquire the MUR domains that are essential for MCV LT autoregulation, potentially leading to viral latency and MCC.
Merkel cell carcinoma (MCC) is a rare but highly aggressive cutaneous neuroendocrine carcinoma [1]. Approximately 80% of Merkel cell carcinomas are caused by Merkel cell polyomavirus (MCV or MCPyV), the only known oncogenic human polyomavirus (HPyV) [2]. MCV is ~5.3 kilobase (kb) viral genome that encodes an early gene locus involved in viral replication, and a late gene locus encoding capsid structure proteins. An early region transcript, MCV large T (LT) protein is generated as a result of alternative splicing of the T antigen locus of the early region [3]. MCV LT is a highly phosphorylated protein that recognizes its viral DNA replication origin and forms a double-hexameric helicase complex to initiate replication of the viral genome [4][5].
MCV LT is a spliced transcript comprising 2 exons producing a protein 817 amino acids in size [3]. The N-terminal end of MCV LT (1–70 aa) contains the DnaJ domain encoding the specific HPDKGG sequence responsible for Hsc70 binding (42–47 aa) [4] and a conserved LXCXE motif for retinoblastoma (Rb)-binding which has high homology to other polyomaviruses [3]. Similar to other polyomaviruses, MCV LT antigen contains multiple conserved domains including a nuclear localization signal (NLS) [6], the origin-binding domain (OBD) [4], and the carboxyl terminal (C-terminus) half which contains several critical elements essential for DNA binding, helicase activity and viral replication [4][5]. In contrast to Simian virus 40 (SV40) LT, expression of wild-type MCV LT inhibits cell proliferation [7][8]. Further analysis of the MCV genome clonally integrated into MCV-positive tumor cells revealed mutations that induce premature truncations in the LT antigen [3], resulting in the deletion of the C-terminus growth-inhibitory domain [7][8].
Protein phosphorylation is a reversible posttranslational modification essential for regulating protein function [9]. Previous reports have shown that phosphorylation is a regulatory mechanism associated with T antigen regulation in Murine polyomavirus (MPyV) and SV40, known polyomavirus models [10][11][12]. The phosphorylation of SV40 LT and MPyV LT is essential for regulating LT antigen function, specifically, viral replication [10][13][14]. MCV LT is a phosphoprotein and contains approximately 82 potential phosphorylation sites (p-sites), which are expected to be crucial for modulation of LT protein function [15]. Full-length LT initiates viral replication and induces Ataxia telangiectasia mutated (ATM) and Ataxia-telangiectasia- and Rad3-related (ATR)-mediated DNA damage response (DDR) and cell cycle arrest, leading to cell death [8][16][17]. It was previously observed that MCV LT serine (S) 816 phosphorylation by the ATM kinase stimulates DDR and cell growth inhibition [17]. Interestingly, an alanine (A) substitutional mutation of S816 only partly restores cell growth, suggesting the possibility of a secondary mechanism contributing to this underlying phenotype [17].
Growing evidence indicates that regulation of viral protein stability may be a key determinant for viral pathogenesis and oncogenesis in many human tumor viruses [18][19][20][21]. SV40 LT protein is relatively stable (t1/2 > 36 h), and regulated at lysine (K) 697 by a reversible acetylation reaction [22]. The effect of acetylation on SV40 LT stability is not changed by proteasomal inhibitor, MG132, indicating that lysine 697 acetylation has a predominant role in regulating SV40 LT stability. Moreover, Shimazu et al. described that a substitution mutation of K679 to glutamic acid (Q) reduced SV40 LT stability and cell growth, suggesting that LT stability is closely related to LT-mediated viral and host protein–protein interactions and cell proliferation.
MCV LT contains a unique domain that exhibits no homology to other polyomaviruses [23][24]. This region comprises two fragments: MCV unique region (MUR) 1 (106 aa) and MUR2 (39 aa), which flank the Rb binding motif [23][24]. This domain contains a unique vacuolar sorting protein (Vam6p) binding sequence (W209) [23], and phosphorylated serines (S147, S220, S239) that interact with Skp-Cullin-F-box (SCF) E3 ligases [15]. MCV LT interactions with SCF E3 ligase complexes induce self-destabilization [15], thus, we hypothesized that this domain functions as a negative regulatory element for LT-mediated cell growth. We sought to determine whether MCV LT MUR domain affects LT protein-mediated cell proliferation. Our results show that deletion of the MUR in MCV LT significantly increases MCV LT protein stability and cell proliferation due to loss of SCF E3 ligase interactions. In contrast, the insertion of MCV LT MUR sequences to SV40 LT destabilizes SV40 LT protein by enhancing SCF E3 ligase interactions, resulting in a moderate decrease in cell proliferation. Interestingly, upon deletion of the MUR domain, MCV LT retained its ability to replicate MCV viral origin and regulate LT-mediated MCV early gene transcription.
The gene sequence of MCV LT is highly conserved among all MCV strains harboring an intact LT sequence [3]. We compared the aligned LT amino acid sequences of 14 human polyomaviruses using PLOTCON software (EMBOSS). As previously shown, the Rb-binding motif in MCV LT is located between the two unique regions (MUR1 and MUR2), unlike other polyomaviruses (Figure 1A) [23][24]. Further assessment of these sequences using IUPred2 and ANCHOR2 allowed us to investigate these MUR fragments, and we ascertained that these two regions are intrinsically disordered (IUPred2 analysis), which may contribute to the multi-functionality of LT including protein-protein interactions [25][26][27]. Because SV40 LT protein interactions and functions are well characterized, we compared sequence variations between SV40 LT and MCV LT (Figure 1B).
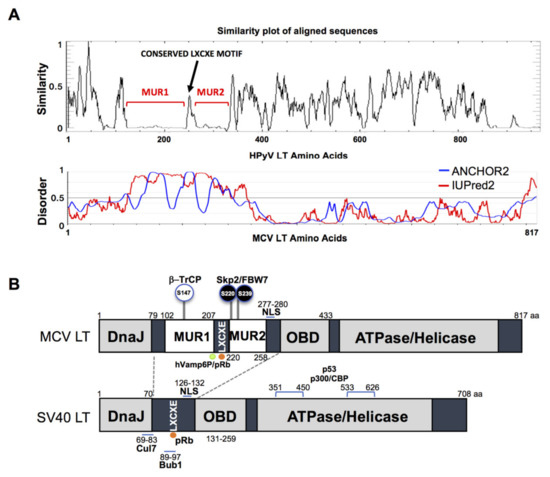
Figure 1. Merkel cell polyomavirus large tumor antigen (MCV LT) similarity and disorder plots. (A) LT amino acid similarity of 14 human polyomaviruses (HPyVs) and disordered region of MCV LT protein. Sliding window plot (6 aa window size) analysis. LT amino acid similarity was compared using PLOTCON (EMBOSS). For disordered region prediction, IUPred2 and ANCHOR2 were utilized to identify disordered protein regions and disordered binding regions in MCV LT, respectively. MCV LT contains a unique disordered region (MUR) divided in two fragments by the conserved LXCXE motif [23][24]. (B) Diagram of MCV LT and SV40 LT domain structures. Compared with SV40 LT, MCV LT has an extended structure, MUR, that serves as an interacting domain with multiple cellular factors. The MUR domain also consists of phosphorylated serines for SCF E3 ligase recognition (phospho-degron motifs) [15].
Previous studies determined that MCV LT MUR domains have multiple E3 ligase binding serines that are not present in SV40 LT [15][28]. To understand the effect of the MUR domains on MCV LT stability, we created a LT MUR deletion mutant (LTdMUR) (Figure 2A) and LT protein stability was analyzed in the presence and absence of the MUR domain using a CHX chase assay. As shown in Figure 2B,C, deletion of the MUR domain significantly reduced LT protein turnover. To further verify if this stability phenotype was MUR-specific, we introduced the MUR sequences to SV40 LT (SV40 LT+MUR) (Figure 2D). Our analysis illustrated that introduction of the MUR mutation to SV40 LT significantly reduced protein stability as shown in Figure 2E. Immunoblot analysis demonstrated a significant reduction in SV40 LT half-life of 36 h [22] to approximately 6 h (Figure 2F).
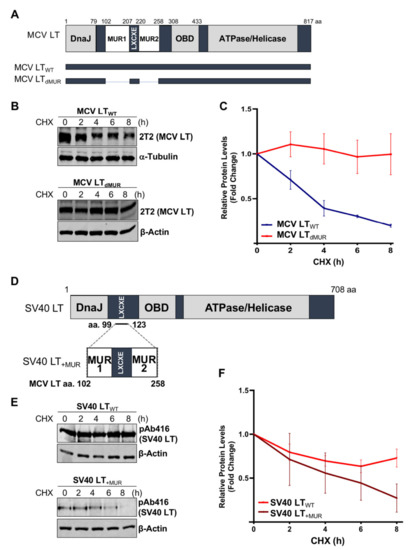
Figure 2. MCV LT unique region (MUR) regulates LT stability. (A) Diagram of MCV LT coding regions and sites of deletion mutations. MCV LT MUR domains: MUR1 (102–207 aa) and MUR2 (220–258 aa) were simultaneously deleted to evaluate LT stability. (B) LT MUR destabilizes LT. LT protein turnover was measured by a cycloheximide (CHX) chase assay using quantitative immunoblot analysis. Cells transfected with LTWT or LTdMUR constructs (0.3 µg and 0.9 µg respectively) were treated with CHX (0.1 mg/mL) 24 h after transfection and harvested at each time point indicated. (C) Protein expression was quantified using a LI-COR IR imaging system. Deletion of the MUR extended the half-life of LT from ~3–4 h up to >8 h. Error bars represent SEM and were calculated using GraphPad Prism software. Data were analyzed using three biological replicates per experiment, n = 3. (D) Diagram of SV40 LT coding regions and sites of insertion mutations. SV40 LT amino acids (99–123) were deleted, and the MCV LT complete MUR domain (102–258 aa) was inserted. (E) MCV LT MUR destabilizes SV40 LT. SV40 LT protein turnover was assessed by a CHX chase assay using quantitative immunoblot analysis. 293 cells transfected with either wild-type SV40 LT (SV40 LTWT) (0.3 µg) or SV40 LT+MUR (0.6 µg) were treated with CHX (0.1 mg/mL) 24 h after transfection and harvested at each time point indicated. (F) Protein expression was quantified using the laser-scanning Odyssey CLX (LI-COR) infrared (IR) imaging system. MCV MUR insertion into SV40 LT reduced SV40 LT turnover to ~6 h. Error bars represent standard errors of the mean (SEM) and were calculated using GraphPad Prism software. Data were analyzed using three biological replicates per experiment, n = 3.





