1000/1000
Hot
Most Recent
 +1 point
+1 point
Cellular therapy has emerged as an attractive option for the treatment of cancer, and adoptive transfer of chimeric antigen receptor (CAR) expressing T cells has gained FDA approval in hematologic malignancy. However, limited efficacy was observed using CAR-T therapy in solid tumors. Natural killer (NK) cells are crucial for tumor surveillance and exhibit potent killing capacity of aberrant cells in an antigen-independent manner. Adoptive transfer of unmodified allogeneic or autologous NK cells has shown limited clinical benefit due to factors including low cell number, low cytotoxicity and failure to migrate to tumor sites. To address these problems, immortalized and autologous NK cells have been genetically engineered to express high affinity receptors (CD16), CARs directed against surface proteins (PD-L1, CD19, Her2, etc.) and endogenous cytokines (IL-2 and IL-15) that are crucial for NK cell survival and cytotoxicity, with positive outcomes reported by several groups both preclinically and clinically. With a multitude of NK cell-based therapies currently in clinic trials, it is likely they will play a crucial role in next-generation cell therapy-based treatment. In this review, we will highlight the recent advances and limitations of allogeneic, autologous and genetically enhanced NK cells used in adoptive cell therapy.
Harnessing the immune system for cancer treatment is one of the most exciting therapeutic possibilities in the history of cancer treatment, and one of the oldest. While the idea of activating the immune system through outside agents to boost an anti-cancer response was first applied in the late 1800s, it has taken a century for similar findings to be validated and applied in contemporary medicine. Modern immunotherapy comprises two broad strategies: treating patients with therapeutic agents that are capable of engaging, expanding and enabling autologous immune cells within the body, and directly modifying effector immune cells to promote their cytolytic ability. Direct cellular modification can take place within the patient through in vivo gene therapy techniques (although no treatment using these methods has gained FDA approval), or by isolating the target cell population and manipulating it ex vivo.
Ex vivo manipulation of immune cells has gained renown through the development and application of chimeric antigen-receptor T cells (CAR-T). Through this process, T cells harvested from a healthy donor (allogeneic) or from the patient (autologous) are expanded and genetically modified ex vivo and re-introduced into that patient [1]. Two CAR-T therapies have been granted FDA approval as of this writing: tisagenlecleucel in relapsed/refractory B cell precursor acute lymphoblastic leukemia (ALL) [2], and axicabtagene ciloleucel in relapsed/refractory diffuse large B cell lymphoma [3], both specific for the B cell antigen CD19. While these treatments have gained success and there are a multitude of ongoing clinical trials using CAR-T, rates of long-term progression-free survival for CAR-T patients are low, frequently attributed to low CAR-T cell persistence in vivo and tumor-associated antigen (TAA) modulation or loss (reviewed here: [4]). One strategy to circumvent antigen escape is through the use of a cytolytic cell that functions independently of antigen: the natural killer (NK) cell.
Natural killer cells are large granular lymphocytes that make up approximately 10–15% of the peripheral blood lymphocyte population, provide a rapid response to viral infection and participate in anti-tumor immune surveillance. In contrast to T cells, NK cell anti-tumor activity does not require antigen recognition in complex with MHC; instead, it is activated following a lack of recognition of “self” markers on tumor cells coupled with a combination of competing activating and inhibitory receptors. There are three major classes of NK receptors: killer immunoglobulin-like receptors (KIRs), the primary MHC-I receptors, C-type lectin receptors that recognize non-classical MHC-I or MHC-I-like molecules, and the natural cytotoxicity receptors. Through the process of NK cell education, the expression of different activating or inhibitory receptors is tuned to prevent NK cells from targeting the body’s own cells while promoting their recognition of non-self cells. Several models of how NK education occurs include licensing/arming, disarming, rheostat and tuning. While the full mechanisms of NK cell education are beyond this article, they have been well reviewed by other authors [5]. Mature NK cells are capable of rapidly recognizing the difference between self and non-self cells. Under healthy conditions, inhibitory NK receptors recognize MHC on the target cells and prevent a cytotoxic response. When a cell is infected with virus or is transformed, downregulation of MHC proteins prevents this inhibitory reaction, and NK cells are activated and subsequently lyse the target cell.
NK cells have two major cytotoxic mechanisms, granulocyte apoptosis mediated by perforin and granzyme and antibody-dependent cell-mediated cytotoxicity (ADCC). Upon NK cell activation, granulocytes are exocytosed, allowing granulocytic perforin to form pores in the cell membrane of target cells [6]. Following cell membrane disruption, granzyme serine proteases are delivered to the cytoplasm of the cell, where they induce apoptosis.
NK cells also induce ADCC, a mode of cell death in which the cell surface receptors Fc?RIIC (CD32c) and Fc?RIIIA (CD16a) bind the Fc portion of antibodies which have bound target antigen. After engaging the Fc receptor, NK cells can induce cell death through granulocytic apoptosis, engagement of tumor-necrosis factor death receptor signaling (mediated by TRAIL proteins) and release of pro-inflammatory cytokines to help generate an adaptive immune response and promote lysis by additional immune cells [7]. Different antibody isotypes generate various degrees of ADCC, allowing this mechanism to be coopted by certain therapeutic antibodies. For example, if a therapeutic antibody is an IgG1 or IgG3, it can bind the Fc? receptors on NK cells, and mediate strong ADCC activity [8]. In addition to serving as one mechanism for antibody cytotoxicity, it is also a potential strategy for additive therapeutic benefit between monoclonal antibodies (mAbs) and cell therapies using NK cells.
The earliest work promoting the NK cell-mediated innate immune response as a tumor therapy focused on the patient’s endogenous NK cell population. These strategies include activating endogenous cells, as well as sensitizing tumor cells to NK-mediated killing. Three general strategies have been developed to promote NK cell-mediated tumor lysis: (1) blocking inhibitory NK cell receptors to increase cytotoxicity against tumors that do not downregulate MHC/HLA proteins, (2) treating patients with NK cell-activating cytokines to promote their expansion, activation and cytotoxicity, and (3) treating patients with additional therapies that result in immunogenic modulation, sensitizing the tumor to NK cell killing.
Similar to checkpoint blockade antibodies, therapeutic mAbs have been designed to block inhibitory receptors on NK cells. Several inhibitory receptor blocking antibodies are currently in clinical trials; however, it is a novel strategy that has yet to come to fruition. Two blocking antibodies are being investigated that interact with the receptor KIR2D. One, IPH2101, was demonstrated to be effective in vitro and in vivo, where it enhanced NK-mediated killing of KIR matched tumor cell lines and enhanced ADCC [9][10]. However, while it was demonstrated to be safe in a Phase I clinical trial of patients with smoldering multiple myeloma, it was not found to be effective in a follow-up Phase II trial [11]. Further studies are ongoing using IPH2101 in combination with lenalidomide, a multiple myeloma medication that has been shown to expand NK cell populations [9]. Similar to IPH2101, a second KIR2D mAb, IPH2102, was demonstrated to be safe in a Phase I trial of hematologic and solid cancers, but no clinical efficacy was observed as a monotherapy in a Phase II trial [12]. Furthermore, a mAb specific for the inhibitory receptor NKG2A increased NK cell killing of leukemia in vivo [13] and was found safe in a Phase I trial in gynecologic cancers [14].
In addition to NK-specific inhibitory receptors, NK cells also express classical checkpoint receptors, including CTLA-4, PD-1, TIGIT, LAG-3 and TIM-3. Inhibition of these negative signaling regulators using checkpoint blockade antibodies may therefore increase their activation state and cytolytic abilities [15]. Numerous clinical trials are underway investigating the effects of checkpoint inhibitors on NK cells, and have been reviewed elsewhere .
Treatment with exogenous cytokines is one of the most frequent immunotherapeutic strategies, with numerous agents being investigated for clinical use in multiple indications and combinations. Interleukins -2, -12, -15, -18 and -21 have been shown to promote NK cytotoxicity and proliferation in vitro and in vivo [16], with IL-2 approved for use in metastatic renal cell carcinoma and metastatic melanoma [17], and approved as a monotherapy in melanoma treatment [18]. Cytokines also play an important role in combination with other immunotherapies and in the activation of allogeneic NK cells. Additionally, several novel immunocytokines are currently in development that promise improved cytokine specificity and activity. The IL-15 superagonist N-803 promotes enhanced NK cell function in vitro and in vivo [19] and has demonstrated safety in early stage clinical trials [20]. Phase I and II trials are currently underway in multiple cancer indications in combination with numerous immuno-oncology agents. Furthermore, NHS-IL-12 is a fusion of IL-12 molecules to a tumor necrosis-targeting human IgG1 that is currently in Phase I and II combination therapy trials; however, its ability to promote NK cell activity is still under investigation [21].
It has been previously reported that certain standard-of-care therapies are capable of changing tumor cell phenotype to sensitize them to killing by cytotoxic immune cells including T cells and NK cells [22][23]. This process has been demonstrated with chemotherapies [24][25][26], endocrine deprivation [27][28], sublethal doses of radiation [29][30][31][32], as well as cytotoxic small molecules, including but not limited to trebananib, ARI-4175, olaparib and bortezomib [33][34][35][36]. These findings present a rationale for combining standard-of-care therapies with a variety of adoptive cell transfer strategies.
NK cells can be obtained from a variety of sources within a patient or healthy donor. While mature NK cells can be isolated from the blood, they can also be differentiated from stem cells, including those isolated from umbilical cord blood, embryonic stem cells and induced pluripotent stem cells. NK cells make up approximately 10–15% of peripheral blood lymphocytes. To obtain clinical grade NK cells, leukapheresis products are first sorted for CD3−CD56+ cell populations, which are then expanded through cytokine treatment, most commonly IL-2 and IL-15 [37][38]. In addition to expanding the NK cell population, these cytokines also activate NK cells, resulting in increased cytotoxicity [16]. Coculture of isolated NK cells with irradiated feeder cell populations has also proven effective and was demonstrated to be safe in a Phase I clinical trial in advanced digestive cancer, and therefore may be a more efficient method for rapid NK cell expansion [39].
NK cells isolated from stem cells benefit from increased ease of storage and purity; however, functional differences from peripheral blood mononuclear cell (PBMC)-derived NK cells have been reported. Notably, in comparison to PBMC-derived NK cells, cord blood NK cells lack expression of the activation marker CD57, have higher expression of the inhibitory receptor NKG2A and express fewer inhibitory KIRs [40], leading to decreased cytotoxicity. However, similar to PBMC isolated NK cells, treatment with IL-2, IL-15, IL-7 or feeder cells has been demonstrated to increase their cytotoxicity [41], indicating that cord blood NK cells remain a viable option [42]. Similarly, a multitude of protocols exist for the differentiation of NK cells from embryonic or pluripotent stem cells that rely on treatment with cytokines or feeder cells [43][44]. Stem cell-derived NK cells may be advantageous over their cord blood counterparts due to their similar cytotoxicity profile in comparison to those isolated from PBMCs [45] and the ability to be maintained as a renewable source of cells [46].
In the clinic, adoptive cell transfer of NK cells can use either autologous NK cells (Figure 1A), isolated or generated from the patient’s own blood or stem cells, or allogeneic NK cells (Figure 1B), in which the NK cells are obtained from a healthy donor.
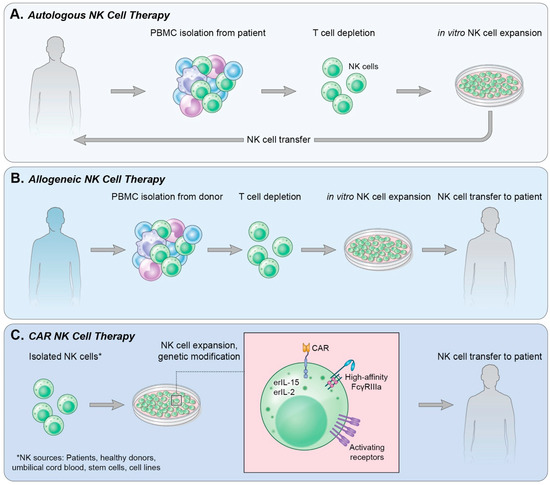
Figure 1. Methods of NK cell therapy. (A) Autologous NK cell therapy uses a patient’s own endogenous NK cells isolated from peripheral blood. T cells are depleted from the culture followed by in vitro expansion of NK cells and reinfusion into the patient. (B) Allogeneic NK cell therapy isolates NK cells from a healthy donor’s PBMCs in a process similar to that in (A); however, following NK cell expansion in vitro, the cells are infused into a patient. (C) CAR NK therapy can use NK cells derived from various sources. NK cells are then genetically modified to express receptors and cytokines of choice and expanded. The CAR NK cells are then infused into a patient.
Notable concerns associated with the use of CAR-therapy, T or NK, are the loss of or decrease in expression of targeted TAAs, rendering CAR-T ineffective and returning CAR-NK cytotoxic function to baseline physiologic capacity of native NK cells, and the influence of the microenvironment on CAR-NK (Figure 2). Several research groups are exploring different mechanisms of combating these concerns. Effectively countering antigen loss (Figure 2, middle panel) and the immunosuppressive TME (Figure 2, right panel) would increase the therapeutic lifespan and efficacy of CAR-NK treatment strategies and may be the backbone of next-next-generation CAR-NK therapy.
To avoid antigen escape, Mitwasi and colleagues developed a universal CAR platform (UniCAR) with an on/off switch to improve safety and controllability [47]. This technology was first demonstrated with CAR-T cells specific for E5B9, a peptide epitope from the nuclear antigen La-SS/B. Because this protein is not found on the cell surface, the CAR-T must be directed to the tumor through the use of a bispecific component, called a target module (TM). TMs usually exist as an E5B9 peptide epitope fused to a scFV of an antibody directed against a TAA. TMs that exist on this platform may be combined with additional TMs to target multiple TAAs simultaneously, inducing heterogeneity in the peptides the CARs are responding to without the risk of off-target effects (Figure 2, middle panel). Mitwasi et al. [47] generated a UniCAR-expressing NK-92 cell line and a TM where the E5B9 epitope is connected to an anti-GD2 mAb on an IgG4 backbone. Although the IgG4 isotype significantly increases the half-life and persistence in vivo, the concern about an on-target/off-tumor effect remains low since NK-92 cells are irradiated prior to infusion. The IgG4 backbone was also chosen because it weakly activates complement C1q, reducing the ability to trigger ADCC in comparison to IgG1 and IgG3. GD2 is highly overexpressed in a variety of human tumors, and one of the only immunotherapy targets in neuroblastoma [48][49]. In fact, a mAb against GD2, dinutuximab, was approved for patients with high risk neuroblastoma [50]. UniCAR NK-92 targeted to GD2-expressing neuroblastoma and melanoma cells can lyse and secrete IFNγ in vitro, as well as antigen-specific lysis of Panc-89 cells directed against EGFR expressed on the cell surface. These data demonstrate proof of concept for further preclinical and clinical experimentation.
Another challenge facing CAR therapy is the TME (Figure 2, right panel). It is as of yet unknown whether CAR-NK cells can effectively traffic to a solid tumor, and whether they are as susceptible to the immunosuppressive TME as CAR-T cells. To avoid the immunosuppressive TME, Wang et al. constructed NK-92 cells expressing a modified CAR consisting of extracellular TGFβRII fused to the intracellular signaling domain of the activating receptor, NKG2D (TGFβRII-NKG2D, termed NK-92-TN) [51]. This fusion CAR, in essence, switches the signal delivered from TGFβ generated from the immunosuppressive TME into an activating signal. In vitro, NK-92-TN cells are resistant to TGFβ suppression, exhibiting no NKG2D downregulation and display increased killing capacity and IFNγ production following TGFβ co-culture. Further interrogation demonstrated that treatment of NK-92-TN with TGFβ did not result in canonical TGFβR signaling, as evidenced by the absence of pSMAD2. In a transwell assay, NK-92-TN cells demonstrate increased migration to tumor cells expressing TGFβ, and have increased expression of chemokine receptors (CCR3, CCR6, CXCR4 and CX3CR1) in comparison to vector control NK-92 cells. In fact, the presence of TGFβ led to a further enhancement of NK-92-TN cytolytic properties, chemoattraction to tumor cells and prevention of CD4 differentiation to Tregs in a hepatocellular carcinoma xenograft model. Although the in vitro data look promising, in vivo, NK-92-TN anti-tumor activity was moderate at best, with a minimal reduction of tumor weight at endpoint, and a slight, albeit significant increase in tumor infiltrating lymphocytes in animals treated with NK-92-TN. These results could be due to the model being used (athymic nude Balb/c mice inoculated with SMMC7721) or that only one dose was administered. Use of humanized mouse models and repeated injections may more closely mirror what is hoped to be observed in the clinic and include the effects these cells could have on the immune populations surrounding them.
Harnessing cells of the immune system to fight various malignancies has been widely explored, with the employment of immunotherapy revolutionizing cancer treatment. However, only a fraction of patients achieve durable clinical responses. Adoptive cell therapy with T cells has undergone extensive research and demonstrated clinical efficacy, and a growing body of evidence suggests that NK cells are also safe and efficacious. While decades of research have demonstrated the difficulties surrounding adoptive cell transfer of allogeneic and autologous NK cells, the data have also found that NK cells represent a pool of innate immune cells poised for rapid tumor clearance. The development of novel, immortalized NK cell lines for off-the-shelf therapy, as well as new gene-editing techniques to arm NK cells with additional mechanisms for targeting tumor cells (CAR) and increasing their cytotoxic potential (endogenous cytokine production) are pushing the field forward in exciting and important ways. It is clear that NK cell transfer is a powerful weapon in the fight against cancer, and that NK cells will play a significant role in the future of immuno-oncology clinical strategies.





