1000/1000
Hot
Most Recent
 +1 point
+1 point
Activities of xenobiotic-metabolizing enzymes have been measured with various in vitro and in vivo methods, such as spectrophotometric, fluorometric, mass spectrometric and radioactivity-based techniques. In fluorescence-based assays, the reaction produces a fluorescent product from a nonfluorescent substrate or vice versa. We describe here historical highlights and current use of (pro)fluorescent coumarin derivatives in evaluating activities of the major types of xenobiotic-metabolizing enzyme systems. Traditionally coumarin substrates have been used to measure oxidative activities of cytochrome P450 (CYP) enzymes. For this purpose, profluorescent coumarins are very sensitive, but generally lack selectivity for individual CYP forms. The entry describes several new coumarin-based substrates for measuring activities of CYP and conjugating enzymes with improved selectivity.
In fluorescence-based assays, the reaction produces a fluorescent product from a nonfluorescent substrate or vice versa. Fluorescence-based enzyme assays are usually highly sensitive and specific, allowing measurements on small specimens of tissues with low enzyme activities. Fluorescence assays are also amenable to miniaturization of the reaction mixtures and can thus be done in high throughput. 7-Hydroxycoumarin and its derivatives are widely used as fluorophores due to their desirable photophysical properties. They possess a large π-π conjugated system with electron-rich and charge transfer properties. This conjugated structure leads to applications of 7-hydroxycoumarins as fluorescent sensors for biological activities.
The metabolism of xenobiotics (foreign substances) in a living organism is vitally important to maintain homeostasis. Xenobiotics are transformed in enzyme-catalyzed oxidation, reduction, hydrolysis, and conjugation reactions to more hydrophilic and excretable metabolites. Usually these reactions yield metabolites with diminished biological activity, but sometimes reactive and more toxic intermediates are formed. In conjugation reactions, a cofactor (small endogenous molecule) is transferred to a functional group of a xenobiotic. The functional group is either already present or is created by reactions of oxidation, reduction or hydrolysis. Conjugations are highly important in overall metabolism because they usually inactivate xenobiotics or make reactive metabolites less harmful. In addition, conjugates are often substrates to transporters mediating excretion in the kidney and liver [1][2].
Cytochrome P450 (CYP) monooxygenases are the most versatile xenobiotic-metabolizing enzymes (XMEs), which oxidize all kinds of lipophilic agents entering the body. CYP-mediated metabolism is an essential mechanism of elimination for both xenobiotics and lipophilic endogenous compounds (endobiotics) such as steroid hormones, bile acids, and lipid-soluble vitamins. CYP enzymes are supported by NADPH CYP reductase, which provides electrons to activate oxygen in the catalytic oxidation reactions of xenobiotics. In all nature, CYP enzymes comprise a very large superfamily, with 18 families in mammals and 57 individual forms in the human genome. Of these CYP forms, ~10 members in families CYP1, CYP2 and CYP3 catalyze the oxidation of xenobiotics to a significant degree. CYPs are most abundantly expressed in the liver, but they are also present in other barrier tissues such as the gastrointestinal tract, lung, skin, nose epithelium, and kidney [3][4][5].
It is estimated that >90% of enzymatic reactions of all organic compounds (general chemicals, natural and physiological compounds, and drugs) are catalyzed by CYP enzymes. About 75% of CYP reactions of clinically used drugs can be accounted for by a set of five hepatic CYP forms: CYP1A2, CYP2C9, CYP2C19, CYP2D6, and CYP3A4 [6]. Conjugation reactions, especially glucuronidation by uridine diphosphate glucuronosyltransferases (UGTs) and sulfonation by sulfotransferases (SULTs), also play a major role in the elimination of xenobiotics and harmful endobiotics [7]. Rates of metabolic enzymes are modulated by numerous endogenous and exogenous factors, including genetic polymorphisms, sex, age, ethnicity, general health conditions, and induction and inhibition by xenobiotics [4][8].
When new chemical entities, especially drugs, biocides, and food additives, are developed, it is essential to evaluate their metabolic pathways in target species and the major XMEs involved. Especially for drugs it is necessary to establish the kinetics and possible inhibition and induction of the XMEs involved.
After performing an in vitro enzyme reaction, formation of metabolites or decrease of substrate can be detected by absorption of light (spectrophotometry), emitted fluorescence (fluorometry), mass/charge ratio (mass spectrometry, MS) or light pulses produced by a radioactive label. For the measurement of a substrate or its metabolites, the most versatile analytical approaches today are liquid chromatography (LC)-MS methods. The drawback of these methods is that the equipment is expensive, their use is labor intensive, and capacity may be saturated [9][10].
Fluorescence is a phenomenon where a molecule (fluorophore) absorbs and then emits lower-energy light. Fluorophores usually contain several conjugated aromatic groups or cyclic molecules with several π bonds or nonbonding electrons. The key parameters in this process are the absorption maxima (λmax) (excitation) and the extinction coefficient at λmax (ε, emission). Loss of energy occurs in excitation due to rapid relaxation to the first singlet excited state (S1) and reorganization of solvent molecules around the altered dipole of the excited state. Fluorescence arises when this excited state is relaxed to the ground state (S0) by emission of a photon. Each fluorophore has a characteristic pair of excitation (λmax) and emission maximum (λem). Chemiluminescence is similar to fluorescence but differs from it in that the electronic excited state is the product of a chemical reaction rather than of the absorption of a photon [11][12].
Fluorescence methods are widely used to evaluate rates and kinetic mechanisms of enzyme reactions. Fluorophore-based enzyme assays are usually highly sensitive and specific. This is particularly valuable when performed on small specimens of tissues or other sources with low enzyme activities. Fluorescence assays are also amenable to miniaturization of the reaction setup and high throughput. Thus, further increases in sensitivity can be achieved with a decrease in demand for enzyme source and fluorescent substrate [13][14][15]. An advantage of fluorophore-based enzyme assays is also that both fixed time and online continuous assays (milliseconds to hours) can be applied for multiple purposes.
Of the various fluorophore-based approaches, we focus here on those that measure fluorescence change of coumarin substrates in reactions catalyzed by several types of XMEs, with main emphasis on our recent experience. In these reactions, a nonfluorescent coumarin derivative is metabolized to corresponding fluorescent 7-hydroxycoumarin metabolite(s) or vice versa (Figure 1). Other fluorophores commonly used for determining activities of XMEs include alkoxyresorufins [16], quinolines [17], furanones [14][17] and some proprietary ones such as Vivid® fluorogenic substrates [18].
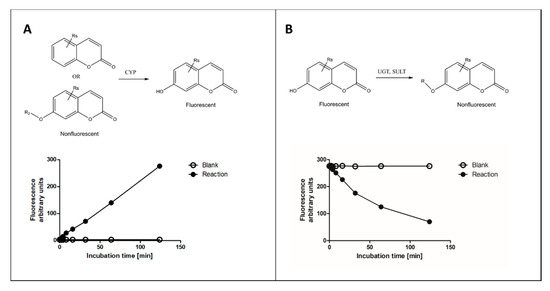
Figure 1. Principles of fluorescence-based cytochrome P450 (CYP) oxidation and conjugation assays with coumarin derivatives. In CYP oxidation, a nonfluorescent coumarin is oxidized to corresponding fluorescent 7-hydroxycoumarin (panel A), which can be conjugated to a nonfluorescent glucuronide or sulfate by uridine diphosphate glucuronosyltransferase (UGT) or sulfotransferase (SULT) enzymes, respectively (panel B). The activities of enzymes can be determined from fixed time (end-point) or continuous assay (kinetic) setups.
The coumarin (2H-1-benzopyran-2-one) scaffold is made of fused benzene and α-pyrone rings. Coumarins can be grouped in six types based on their extended backbone structures (Figure 2): (1) simple coumarins, (2) furanocoumarins, (3) pyranocoumarins, (4) sesquiterpenyl coumarins, (5) oligomeric coumarins and (6) miscellaneous coumarins [19]. The coumarin scaffold occurs in constituents of food and numerous natural and synthetic products such as drugs, perfumes, rodenticides, and laser dyes [19][20].
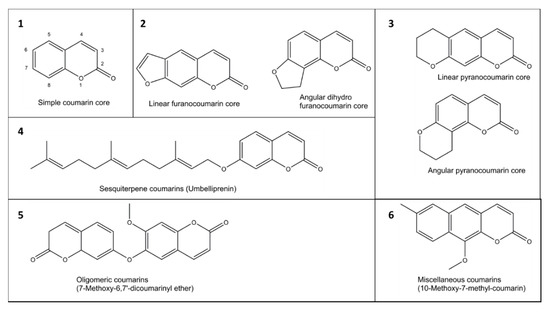
Figure 2. Examples of six groups (1–6) of coumarins in plants.
Coumarin is widely used as a fluorophore due to its desirable photophysical properties. Coumarin possesses a large π-π conjugated system with electron-rich and charge transfer properties. This conjugated structure leads to their applications as fluorescent sensors for biological activities. Substitution at the 7-position with electron-donating groups yields highly fluorescent molecules, 7-hydroxycoumarin (umbelliferone) being the core structure of these molecules. Many suitable combinations of substitutions at the 7-hydroxycoumarin scaffold do not destroy the intense fluorescence [11][21]. However, we have noticed that 4, 5-dialkyl substituents suppress the fluorescence intensity to a minimum (unpublished data), and fluorescence of 7-hydroxywarfarin is low. Coumarin and many of its derivatives are converted to 7-hydroxycoumarin metabolites in the typical oxidation reaction of CYP enzymes (Figure 1).
The fluorescent properties of 7-hydroxycoumarin were already used in the 1950s to evaluate its metabolism in the laboratory of R.T. Williams. It was observed that, although fluorescence of 7-hydroxycoumarin was strong in ultraviolet light at pH ~10, its glucuronide and sulfate conjugates showed little or no fluorescence. Based on this, a sensitive fluorescence method was developed for the determination of β-glucuronidase activity, in which nonfluorescent glucuronide conjugate was hydrolyzed to fluorescent 7-hydroxycoumarin [22]. An in vitro method for measuring coumarin 7-hydroxylation activity using fluorescence detection was subsequently developed for CYP enzymes in the same laboratory. This study also confirmed the marked interspecies differences in hepatic coumarin 7-hydroxylation capacity [23]. A simplified version of the 7-ethoxycoumarin O-deethylation to 7-hydroxycoumarin assay was published by Aitio and colleagues in the late 1970s [24]. This method became popular and is still being used in fixed time (end-point) enzyme assays.
Numerous new 7-substituted coumarin derivatives for measuring activities of CYP enzymes have been introduced since the 1970s. These include 7-ethoxycoumarin, 3-cyano-7-ethoxycoumarin (CEC), 7-ethoxy-4-trifluoromethylcoumarin (EFC), 7-methoxy-4-trifluoromethylcoumarin (MFC), 7-methoxy-4-aminomethylcoumarin (MAMC), 3-[2-(N,N-diethyl-N-methylammonium)ethyl]- 7-methoxy-4-methylcoumarin (AMMC), and 7-benzyloxy-4-trifluoromethylcoumarin (BFC) (Figure 3). These alkylated derivatives of 7-hydroxycoumarin are ‘blocked’ substrates, whose fluorescence signal is minimal compared to the 7-O-dealkylated oxidation product. Some of these have been commercialized for use in CYP-mediated drug interaction studies, pioneered by the Gentest Corporation (now Corning) [13][17][25]. A major shortcoming of these coumarin derivatives is that, with few exceptions, they are not selective but oxidized by several human CYP forms [13][17][25][26][27]. Coumarin itself is an example of a highly selective CYP substrate, as it is oxidized practically exclusively by human CYP2A6 to fluorescent 7-hydroxycoumarin [28][29]. In addition to being excellent substrates, numerous coumarins are potent CYP inhibitors, particularly furanocoumarins such as methoxsalen [30][31][32].
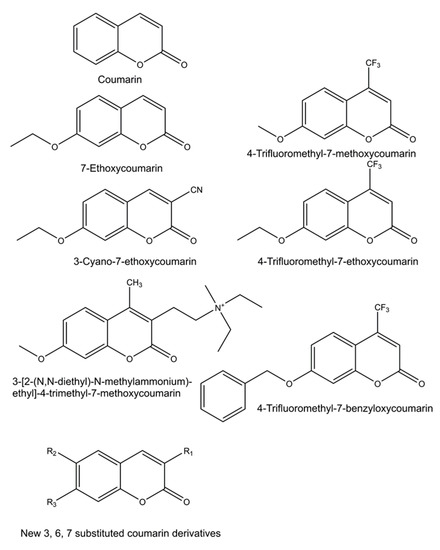
Figure 3. Structures of profluorescent CYP substrates oxidized to fluorescent 7-hydroxycoumarin derivatives. New coumarin derivatives can have various combinations of substituents at position 3 (3-hydroxyphenyl, 3-methoxyphenyl, 3-benzyloxo, 4-fluorophenyl, 4-trifluoromethylphenyl or pyridyl); at position 6 (hydrogen, methyl, methoxy, chloro or hydroxy); and at position 7 (hydrogen or methoxy).
Ideally, sensitive and selective fluorophore substrates for different forms of XMEs would provide effective and low-cost tools for studying form-specific metabolism in simple assays using all kinds of enzyme sources, including whole tissue or subcellular fractions. However, although many fluorophore substrates are available, new substrates are needed as most human XMEs are still missing the optimal substrate, especially regarding selectivity for different enzyme forms. The flexible chemistry of the coumarin core offers diverse options for functionalization to design novel fluorescent substrates.
As part of an effort to find novel coumarin-based, pharmacologically active molecules, we have synthesized multiple derivatives of the coumarin scaffold. The synthesis protocols were kept simple and robust, and inexpensive starting materials were used. The detailed protocols are published [33][34][35]. In a typical procedure, a mixture of a salicylaldehyde derivative, a phenylacetic acid derivative, acetic acid anhydride and trimethylamine is heated at 100°C. After cooling, NaHCO3 solution is added, and the precipitate is filtered, dried, and recrystallized. The acetyl group(s) are removed by treating the compound with methanol/NaOH solution. The solution is acidified with HCl, and the precipitate is filtered and recrystallized if needed. Based on the elemental analysis and/or 1H-NMR the purity of compounds is typically >95%.
When setting up fluorescence assays to quantify enzyme activities, the first step is to establish incubation conditions supporting the reaction. Phosphate or Tris-HCl buffers are commonly used to gain the optimal ion concentration and pH of 7.4. All enzymes require their cofactors: NADPH for CYP oxidations, uridine 5’-diphosphoglucuronic acid (UDPGA) for UGTs, 3’-phosphoadenylyl sulfate (PAPS) for SULTs and S-adenosylmethionine (SAM) for COMT. The fluorescent substrates must have sufficient aqueous solubility (at least µM range), efficient metabolite formation, low background fluorescence, high signal-to-noise ratio, and appropriate excitation and emission wavelengths in the UV range [12][36].
During incubation a metabolite is formed, and the amount of substrate is decreased. Fluorescent 7-hydroxycoumarins are excellent for measuring substrate decrease, because several conjugating enzymes transfer a substituent to the 7-hydroxyl group making the product nonfluorescent (Figure 1B). A practical advantage of measuring conjugating reactions is that 7-hydroxycoumarins act as standards, allowing for reliable and accurate quantification of the reaction, whereas CYP oxidation reactions require the availability of a selective reaction standard for exact quantitation. However, in the latter case surrogate standards such as 7-hydroxycoumarin can be applied.
The solubility of coumarin in water is very high (20 mM). Lipophilic substituents decrease solubility, causing much variability to solubility among coumarin derivatives. The solubility of 7-hydroxycoumarins is higher than that of the corresponding derivatives. In our experience, even the poorest soluble coumarin derivatives have been soluble at µM concentrations. We routinely use 0.05–10 µM concentrations in the assays. Enzyme sources (especially tissues) and cofactors can produce background fluorescence. Contamination of tissue samples with blood causes problems because hemoglobin absorbs light efficiently at 370–450 nm. Too high concentrations of enzyme sample reduce fluorescence due to absorption of excitation or emission light and produce insufficient signal-to-noise ratio in the reaction. In the continuous (kinetic) assays up to 5% tissue in the reaction mixture does not excessively suppress fluorescence. Recombinant or purified enzymes added in small volumes in the incubation mixture yield no increase in the background. Tissue samples can be removed in the fixed time measurements by precipitation, but this adds extra steps. When very low activities are measured in fixed time assays, small differences in composition between sample and negative control may cause background fluorescence even after precipitation. In continuous assays background fluorescence is usually not a major issue, as fluorescence change is monitored against background during the entire measurement period.
Although both the excitation and emission peaks are broad for fluorescent compounds, it is important that these are optimized in the assays. Excitation wavelengths typically range from 300 to 420 nm and emission wavelengths from 350 to 500 nm for 7-hydroxycoumarin derivatives. Coumarins are also fluorescent; however, their excitation wavelengths are lower than 7-hydroxycoumarins. NADPH is strongly fluorescent <390 nm if excitation ~350 nm is used, but it is nonfluorescent at excitation >400 nm. The cofactors UDPGA and PAPS do not cause background fluorescence. Using excitation >400 nm minimizes background fluorescence and gives high enough emission fluorescence at 450–500 nm for coumarin-based assays. Negative control incubations lacking the enzyme, substrate or cofactor are needed to monitor background fluorescence.
When selecting the enzyme source, one must keep in mind that tissues and their cellular subfractions contain several forms of XMEs, most notably the liver. Since almost all fluorescent probes are not selective or lose selectivity at high substrate concentrations, heterologously expressed individual XMEs should be used when enzyme-specific reactions are studied. Nowadays, there is a good commercial supply of expressed human CYP, UGT and SULT enzymes.
To understand an enzyme reaction and its physiological role, it is essential to determine the effect of substrate concentration on the reaction rate (Km—the substrate concentration giving half-maximum rate) and the limiting rate for the reaction (Vmax—the rate at saturating substrate concentration). Experiments can then be rationally planned for evaluating potential inhibitors and characteristics of inhibition, including inhibitor potency and whether the inhibition mechanism is reversible, irreversible, competitive, un-competitive (catalytic) or mixed type of inhibition [12][36]. When interpreting the results, it should be considered that especially CYP3 enzymes may deviate from Michaelis–Menten kinetics with several probes, including fluorescent substrates [37][38]. In summary, sensitivity of fluorescence-based assays consists of the fluorescence properties of the substrates, products, cofactors, amount and purity of samples, and incubation time. All can be optimized as described here.
The UGTs catalyze ~35% of all drug conjugation reactions. They catalyze glucuronidation, i.e., transfer of the glucuronic acid moiety from UDPGA onto nucleophilic hydroxyl, amine, carboxylic acid, thiol or thioacid groups of the substrates. There is a higher concentration of UGTs in the small intestine than in the liver, and expression levels of individual UGTs differ in these tissues [39].
As discussed above, glucuronidation of the C7 hydroxyl group abolishes coumarin fluorescence (Figure 1B). Substituents at positions such as C3 or C4 do not quench the fluorescence of 7-hydroxycoumarin derivatives, but rather modify its intensity, depending on the substituent’s chemical nature (Figure 5). Based on this phenomenon, we developed a convenient quantitative multiwell plate assay to measure the glucuronidation rate of 7-hydroxycoumarin derivatives by human recombinant UGT enzymes. At 20 µM substrate concentration, the most active HFC glucuronidation catalysts were UGT1A10 followed by UGT1A6 >UGT1A7 >UGT2A1, whereas at 300 µM UGT1A6 was ~10 times better a catalyst than the other UGTs [40]. The activities of UGTs 1A3, 1A8, 1A9, 2B4 and 2B7 were low, whereas UGT1A1 and UGT2B17 exhibited no HFC glucuronidation activity.
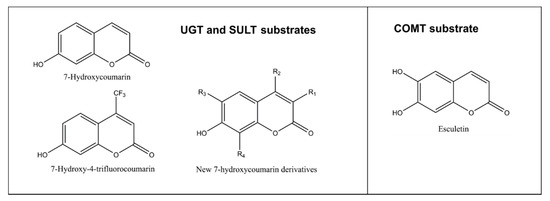
Figure 5. Structures of substrates conjugated to nonfluorescent metabolites. An exception is esculetin, which is weakly fluorescent but its 6-methoxy-7-hydroxycoumarin (scopoletin) is strongly fluorescent. New 7-hydroxycoumarin derivatives can have different combinations of substituents at position 3 (methyl, ethyl, 4-methylphenyl, 4-methoxyphenyl, 4-fluorophenyl, 4-hydroxphenyl, 4-dimethylaminephenyl, pyridine-3-yl or triazole), at position 4 such as methyl or trifluoromethyl, at position 6 such as methoxy or at position 8 such as methyl.
In a follow-up study [33] we constructed predictive homology models for the human UGT1A enzymes to design selective substrates for them. Six novel C3 substituted coumarin derivatives (4-fluorophenyl, 4-hydroxyphenyl, 4-methoxyphenyl, 4-dimethylaminophenyl, 4-methylphenyl or triazole) were synthesized. All tested compounds were glucuronidated to nonfluorescent conjugates by the intestinal UGT1A10. 4-dimethylaminophenyl and triazole C3 substituted 7-hydroxycoumarins were selective substrates for UGT1A10. The hydroxyl group of 7-hydroxycoumarin was oriented toward the cofactor UDPGA in the active site of the UGT1A10 model, indicating that there is space in the active site for an additional 6- or 5-ring substituent at position C3. The extensive fluorescence of these new 7-hydroxcoumarin derivatives as UGT1A10 substrates makes them suitable for quick and convenient activity measurements in a high-throughput format.
In a recent study [41] we used the multiwell plate assay to evaluate the glucuronidation characteristics of several 7-hydroxycoumarin derivatives in intestine and liver microsomes of beagle dog and human. High glucuronidation rates were observed in the liver and small intestine. The results underscored the high capacity of dog intestine for glucuronidation and revealed that different UGT enzymes mediate glucuronidation with distinct substrates selectivity in dog and human, even if all the substrates are 7-hydroxycoumarin derivatives.
Sulfonation by SULT enzymes takes place for nucleophilic phenols, alcohols or primary or secondary amines. The sulfate conjugates are negatively charged and more water-soluble than the parent compounds. These metabolites are readily excreted primarily to urine or sometimes transported to bile. Sulfonation and glucuronidation reactions act in a complementary way. Sulfonation as part of overall xenobiotic metabolism usually protects the body against adverse health effects. However, sulfate conjugates of hydroxylated amines such as heterocyclic amines can form reactive metabolites with detrimental consequences [39][42].
In analogy with glucuronidation, fluorescence is diminished by the sulfonation of 7-hydroxycoumarins (Figure 1B and Figure 5). We developed recently a fluorescence-based kinetic sulfonation assay using this principle, using the same 7-hydroxycoumarin substrates as in the glucuronidation assay described above [43]. Because this assay was sensitive and quantitative, it could be applied to determine sulfonation kinetics in liver cytosol of human, mouse, rat, pig, rabbit, dog and sheep. The assay can also be used to study potential inhibitors of sulfonation to reveal potential drug interactions.
COMT is the selective methylating enzyme of catechol structures, compounds having two hydroxyl substituents in adjacent positions of an aromatic ring. In humans, there is one COMT gene, from which a long membrane-bound and a short cytosolic COMT is expressed. Its activity can be measured with esculetin, a catecholic coumarin derivative [44][45]. The weakly fluorescent esculetin is methylated by COMT to either 6-methoxy-7-hydroxycoumarin (scopoletin) or 6-hydroxy-7-methoxycoumarin (isoscopoletin) (Figure 5).
Isoscopoletin is nonfluorescent, and scopoletin is strongly fluorescent. Therefore, increase of fluorescence takes place in esculetin 6-O-methylation reaction, to which a convenient fluorescent-based assay for COMT activity is based.





