1000/1000
Hot
Most Recent
 +1 point
+1 point
Sphingolipids are an important class of lipids that comprise a fatty acid that is attached to a sphingoid long-chain base. An increasing amount of evidence links these sphingolipids to neurodegenerative disorders.
Neurodegenerative disorders (ND) are progressive disorders that impair specific brain areas or neuronal subtypes, accompanied by a gradual increase in signs and symptoms linked to the affected brain areas. A puzzling observation in ND is that in initial, pre- and early clinical stages, only specific neuronal subtypes degenerate, whereas in later, advanced stages, other neuronal subtypes are affected as well, resulting in more comprehensive neuronal degeneration. For example, in Parkinson’s disease (PD), dopaminergic neurons are initially affected, but first symptoms manifest only when the loss is around 70%, followed by broader neurodegeneration [1]. An early diagnosis and slowing down disease progression are key in the treatment of ND; however, there is at least a partial lack of knowledge about the underlying disease pathogenesis, which is a barrier to the development of targeted drugs. While several ND gene mutations have been identified that are causative, the majority of ND remain sporadic, or in the case of PD, both sporadic and genetic forms of the disease exist. Nevertheless, the existence of these genetic forms has enabled the creation of animal models to understand the underlying mechanisms that are affected in ND. Such studies have strongly contributed to our current knowledge and several common molecular mechanisms have been identified to play an important role in the pathogenesis of ND. One interesting observation is the presence of deposits, including proteins, in, e.g., Alzheimer’s disease (AD) and PD, and iron in PD and neurodegeneration with brain iron accumulation (NBIA) [2][3][4][5][6]. It remains, however, unclear if these deposits are the consequence of upstream cellular defects or rather (partially) the cause of the observed signs. Furthermore, mitochondrial dysfunction is a recurring observation in ND, including defects at the level of the energy production and mitophagy, a mitochondria-specific form of autophagy, which has been linked to several NDs [7][8][9][10][11][12]. More recently, lipids (and more specifically sphingolipids) have been shown to play a crucial role in neurodegeneration.
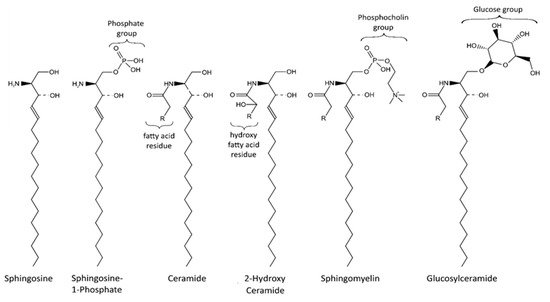
Sphingolipids are an important class of lipids that comprise a fatty acid that is attached to a sphingoid long-chain base ( Figure 1 ). The sphingoid bases are aliphatic amino alcohols [13]. When a fatty acid binds to the amino group, ceramides are formed, which are the base of all sphingolipids. The incorporated fatty acids are very diverse. In addition to different carbon chain lengths, variations in saturation and hydroxylation exist [14]. Furthermore, sphingolipid groups attach different head groups to the first carbon atom of the sphingoid base. This makes the lipids amphipathic, meaning that they have both a hydrophobic and a hydrophilic region. A ceramide that consists of only one hydroxyl head group has a limited hydrophobicity. The amphipathic effect is stronger in more complex sphingolipids such as sphingomyelin and glycosphingolipids that bind phosphatidylcholine and saccharides, respectively ( Figure 1 ) [14]. Ceramide metabolism occurs via three distinct pathways ( Figure 2 ). The first pathway, de novo synthesis, occurs in the endoplasmic reticulum (ER). Here, dihydrosphingosine is formed from serine and a palmitoyl- CoA. Once dihydrosphingosine binds to a fatty acyl CoA, dihydroceramide is formed. This step is catalyzed by ceramide synthases. In humans, six different ceramide synthases are described that use different fatty acids as substrates. The dihydroceramide is subsequently converted into ceramide [15][16]. The second pathway is the salvage pathway, which takes place in the lysosomes. In this pathway, ceramides are obtained by the catabolism of complex sphingolipids. The sphingoid base is formed, which is reacetylated to ceramides [16][17]. The third and most rapid metabolic pathway is sphingomyelin hydrolysis, which occurs in the Golgi apparatus. Sphingomyelin is degraded to ceramide and phosphatidylcholine [16][18]. Ceramide metabolism is tightly regulated by enzyme expression, post-translational modifications, and allosteric mechanisms [14]. Sphingolipids are bioactive lipids involved in numerous cellular processes. The various sphingolipids fulfill different roles and may have antagonistic effects. Ceramide regulates cell stress response and inhibits cell growth, as well as promoting apoptosis, cell senescence, and autophagy, in particular mitophagy [19][20]. Several ceramides have been associated with mitochondrial dysfunction, including reduced mitochondrial respiratory chain, increased reactive oxygen species (ROS), and reduced mitochondrial membrane potential [16]. The ceramide precursor sphingosine has similar functions. In addition to the regulation of actin cytoskeleton and endocytosis, it induces cell cycle arrest and stimulates apoptosis [19][21]. Sphingosine-1-phosphate (S1P), a degradation product of ceramide, exhibits antagonistic effects on ceramide and sphingosine. It promotes cell growth and survival, and prevents apoptosis [14][19][21][22]. Furthermore, it regulates cell migration [19]. The more complex sphingomyelin is part of the myelin sheath that surrounds axons and it stimulates axonal growth and neuronal transmission. Additionally, it is required for presynaptic plasticity and neurotransmitter receptor localization [23][24][25]. Another large and complex group of sphingolipids are glycosphingolipids. They are involved in post-Golgi transport and cell differentiation. Studies have revealed hydroxylated galactosyl ceramides to be essential for myelin structure, function, and maintenance. However, they are not essential for myelin formation [26]. Hydroxy sphingolipids, in general, are lipids of which the fatty acid residue is hydroxylated [27]. Their function has also been linked to the epidermal permeability barrier, axonal development, the cAMP-dependent signaling pathway that regulates the cell cycle, and apoptosis [26][27].
To date, ten genes have been identified to be classic NBIA genes [6]. The most common form of NBIA that occurs in 35–50% of NBIA cases is pantothenate kinase-associated neurodegeneration (PKAN), which arises from mutations in pantothenate kinase 2 ( PANK2 ). The second largest group, constituting 20% of NBIA patients, is PLA2G6-associated neurodegeneration (PLAN) which is caused by mutations in phospholipase A2 ( PLA2G6 ). Mitochondrial membrane protein-associated neurodegeneration (MPAN), induced by mutations in chromosome 19 open reading frame 12 ( C19orf12 ), was identified in 6–10% of NBIA patients, and ß-propeller-associated neurodegeneration (BPAN), caused by a mutation in tryptophan-aspartic-acid repeat domain 45 ( WDR45 ), was identified in 1–2% of NBIA cases [28]. Fatty acid hydroxylase-associated neurodegeneration (FAHN) (with fatty acid 2 hydroxylase ( FA2H ) mutation), neuroferritinopathy (with Ferritin ( FTL1 ) mutation), aceruloplasminemia (with ceruloplasmin ( CP ) mutation), Woodhouse–Sakati syndrome (with DB1 and Cul4-associated factor 17 mutation), Kufor–Rakeb syndrome (with ATPase cation transporting 13A2 mutation), and COASY protein-associated neurodegeneration (with CoA synthase mutation) are rare diseases affecting less than 1% of NBIA cases ( Table 1 ) [28]. In addition, many patients present with typical NBIA symptoms; however, to date, no disease-causing mutations have been identified.
| Disease | Gene | Protein Localization | Pathway |
|---|---|---|---|
| PKAN | PANK2 | Mitochondrium | Coenzyme A Biosynthesis |
| PLAN | PLA2G6 | Mitochondrium, ER and Cytosol | Lipid Metabolism |
| MPAN | C19orf12 | Mitochondrium, ER and MAM | Lipid Metabolism |
| BPAN | WDR45 | ER | Autophagy |
| FAHN | FA2H | ER | Lipid Metabolism |
| Neuroferritinopathy | FTL1 | Cytoplasm | Iron Homeostasis |
| Aceruloplasminemia | CP | Plasma Membrane | Iron Homeostasis |
| WoodHouse–Sakati Syndrome | DCAF17 | Nucleolus | Unknown function |
| Kufor–Rakeb Syndrome | ATP13A2 | Mitochondrium and Lysosome | Autophagy |
| CoPAN | COASY | Mitochondrium and Cytosol | Coenzyme A Biosynthesis |
| PD | SNCA | ER and Golgi Apparatus | Endo-Lysosomal Pathway |
| VPS35 | Retromer Complex | Endo-Lysosomal Pathway | |
| LRRK2 | ER and Golgi Apparatus | Endo-Lysosomal Pathway | |
| DJ-1 | Mitochondrium | Mitochondrial Function | |
| Parkin | Mitochondrium and Cytosol | Mitochondrial Function and Mitophagy | |
| PINK1 | Mitochondrium | Mitochondrial Function and Mitophagy |
PKAN is inherited in an autosomal recessive fashion and can be subdivided into “ classic ” , “ atypical ” and “ intermediate ” types, depending on the age of onset and disease progression [29]. Typical symptoms are dystonia and cognitive dysfunction [29]. PANK2 encodes the enzyme pantothenate kinase 2, which is one of four pantothenate kinase proteins. It is localized to mitochondria where it phosphorylates the coenzyme A precursors pantothenate, N -pantothenoyl-cysteine, and pantetheine [30][31]. An examination of PKAN patient-derived plasma showed elevated lactate levels indicative of mitochondrial dysfunction [32]. This was confirmed by experiments using knock-out (KO) mice, knock-down (KD) Drosophila and iPSC-derived cortical neurons that showed altered mitochondrial membrane potential, swollen mitochondria and increased ROS [33][34][35]. In addition, Coenzyme A, which is directly involved in lipid metabolism, was reduced upon the KD of PANK2 in a fruit fly, which is consistent with the increased pantothenate levels found in the plasma of patients [32][33]. Moreover, experiments were performed to investigate the lipid composition of plasma and red blood cells of PKAN patients. Glycerolipids, the sphingolipid C16:1 and sphingomyelin were reduced. Interestingly, the analysis of the distribution of the different lipids in the membrane of red blood cells showed that the molar percentage of sphingomyelin was increased compared to that of the other lipids [32][36]. Thus, studies suggest an interaction between the mitochondrial PANK2 and altered sphingolipids metabolism. However, the nature of this link and how this results in ND are yet to be clarified.
PLAN is inherited in an autosomal recessive manner. It is an umbrella term for various disorders as a consequence of a plethora of mutations in the PLA2G6 gene [37]. The different PLAN types depend on the age of onset and severity of symptoms and can be divided into classic infantile neuroaxonal dystrophy (INAD), atypical neuroaxonal dystrophy (NAD) and PLA2G6-related dystonia-parkinsonism [29]. However, no direct correlation between the mutations and PLAN types has been identified [38]. Neurological examinations of INAD and atypical NAD patients often reveal cerebellar atrophy, iron deposition in the globus pallidus, abnormal axons (spheroid bodies), and Lewy bodies, as well as hyperphosphorylated tau [39][40][41].
iPLA2ß catalyzes the hydrolysis of glycerophospholipids, suggesting that alterations in phospholipid metabolism make up the underlying molecular mechanism of PLAN. Studies in a KO mouse model indeed showed an altered phospholipid composition in the spinal cord. [42]. However, this could not be identified in a mutant Drosophila model [43]. Studies on lipid alterations mediated by a mutation in iPLA2ß revealed increased lipid peroxidation levels in a KO Drosophila model. Lowering the lipid peroxidation levels rescued the accompanying motor symptoms in a fly model and the mitochondrial membrane potential in patient-derived fibroblasts [44]. Remarkably, the loss of iPLA2ß in flies leads to an accumulation of ceramide. Furthermore, iPLA2-VIA binds to Vps26 and Vps35, two subunits of the retromer complex, which is important for lipid recycling. Hence, the loss of iPLA2-VIA resulted in impaired retromer function, preventing sphingolipids from being transported to the plasma membrane. Instead, sphingolipids are converted to ceramide in lysosomes. This results in ceramide accumulation, which has a toxic effect on the cell [43].





