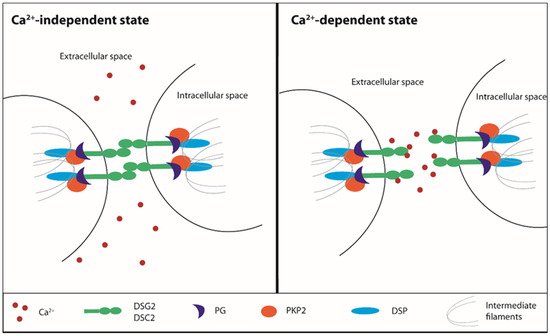
PKP2, encoding for plakophilin-2, is the most frequently mutated gene in ACM patients
[36]. It has also been linked to other inherited cardiac arrhythmia syndromes, such as Brugada syndrome
[51], idiopathic ventricular fibrillation, hypertrophic cardiomyopathy, and dilated cardiomyopathy, even if with a low frequency
[52]. PKP2 is expressed in both myocytes and non-myocytes
[53]. The main function of PKP2 is to guarantee mechanical stability during the desmosomal intermediate filament assembly required for cell-to-cell contact. PKP2 is part of the so-called ‘connexome’
[54]. Different studies highlighted the consequent novel role for PKP2 in the intracellular signaling regulation, electrophysiological and trafficking regulation, and the control of transcription processes
[33].
Data from a cardiomyocyte-specific tamoxifen-activated
Pkp2 homozygous knockout (
Pkp2-cKO) mouse model correlated the lack of PKP2 with transcriptional alteration of Ca
2+ homeostasis
[33]. This investigation reported the development of a cardiomyopathy of RV predominance that become evident at 21 days and then progressed into biventricular cardiomyopathy and HF. Transcriptome analysis showed that the transcripts of proteins involved in maintaining the intracellular Ca
2+ concentration were downregulated in
Pkp2-cKO hearts. The low level of transcripts relevant to Ca
2+ cycling, i.e.,
Ryr2,
Ank2,
Cacna1c, and
Trdn, accompanied a decreased expression of corresponding proteins and the impairment of the EC mechanism
[33]. Accordingly, the downregulation of AnkB (encoded by
Ank2) and triadin (encoded by
Trdn), which contribute to maintaining the structural integrity of dyadic junctions
[55][56], significantly reduced the distance between CaV1.2 and RYR2
[33]. In addition,
Pkp2-cKO-derived cardiomyocytes showed decreased I
Ca,L density and a slower rate of current inactivation, which is consistent with the reduced expression of
Cacna1c [33]. It should, however, be pointed out that, although the peak Ca
2+ current was decreased, the total Ca
2+ charge (i.e., the total amount of Ca
2+ entering the cell upon membrane depolarization) was unaltered as compared to wild-type cardiomyocytes because of the slower inactivation rate
[33]. Notably, the loss of PKP2 was also correlated with a reduction in the SR Ca
2+ leak due to the downregulation of both
Ryr2 and
Casq2 [33] (
Figure 1). As a consequence, the SR Ca
2+ content was remarkably increased in
Pkp2-cKO cardiomyocytes, which exhibited an increase in the amplitude and frequency of spontaneous Ca
2+ release events (due to RYR2 sensitivity to intraluminal Ca
2+ levels) and were therefore more prone to release SR Ca
2+ during the EC coupling
[33]. Accordingly, when
Pkp2-cKO cardiomyocytes were paced at increasing rates, they displayed both early and delayed after-transients, which were sufficient to generate ventricular arrhythmogenic events during β-adrenergic stimulation with isoproterenol
[33]. A more recent investigation focused on the early events driving the remodeling of the Ca
2+ handling machinery in RV-derived PKP2-cKO (
Pkp2-cKO-RV) cardiomyocytes isolated 14 days after tamoxifen injection
[57], i.e., when cardiomyopathy was not evident yet. This report revealed an increase in RyR2-dependent Ca
2+ release and RyR2-mediated Ca
2+ sparks due to the remarkable elevation in the SR Ca
2+ load that was caused by a Cx43-dependent increase in membrane permeability
[57]. Notably, uncoupled Cx43 hemichannels may provide an alternative pathway for extracellular Ca
2+ influx
[58][59][60] and may, therefore, contribute to refilling the SR Ca
2+ store in a SERCA2A-dependent manner. In addition, RyR2’s eagerness to release Ca
2+ may be boosted by the observed phosphorylation in Thr2809, an amino acid residue near the consensus sequence for CaMKII and PKA
[57]. These alterations were not detected in PKP2-cKO LV cardiomyocytes, thereby suggesting that this asymmetric dysregulation of the Ca
2+ handling machinery precedes overt ultrastructural alterations and manifestations of ACM.
The relationship between PKP2 and Ca
2+ machinery has also been highlighted by a recent bioinformatic approach that took advantage of a database containing transcriptomic information from human hearts, searching for coordinated transcription networks that are subjected to variations based on PKP2 abundance. The results were then validated with the information deriving from
Pkp2-cKO murine hearts, thereby confirming the downregulation of
RYR2,
ANK2, and
CACNA1C [61]. The results of the combined data supported the idea of a correlation between the PKP2 expression and the abundance of transcripts related to intracellular Ca
2+ homeostasis. In this context, mathematical modeling confirmed that the PKP2-dependent downregulation of RYR2 and CASQ2 proteins is sufficient to cause the decrease in SR Ca
2+ leak, which results in enhanced SR Ca
2+ loading and EC coupling
[33]. Accordingly, studies carried out on human iPSC-CM from a
PKP2-mutated ACM patient showed that they present an abnormal Ca
2+ handling capacity
[62]. Of note, a recent investigation in iPSC-CM suggested that the Ca
2+ handling machinery could also be affected by
DSG2 mutations. Accordingly, although the systolic and diastolic Ca
2+ levels were similar, human ACM iPSC-CM exhibited spontaneous SR Ca
2+ release and DADs in both the absence and the presence of β-adrenergic stimulation
[63]. This investigation did not evaluate the molecular expression of Ca
2+-related proteins, but further supports the notion that desmosomal mutations may affect the cardiac Ca
2+ toolkit in ACM.
 +1 point
+1 point