Ranaviruses (Iridoviridae), including Frog Virus 3 (FV3), are large dsDNA viruses that cause devastating infections globally in amphibians, fish, and reptiles, and contribute to catastrophic amphibian declines. Our findings imply that Ranaviruses like FV3 have acquired previously unknown molecular mimics, interfering with host IFN signaling during evolution.
1. Introduction
Frog virus 3 (FV3) is a large (~105 kb), double-strand DNA (dsDNA) virus belonging to the Ranaviruses genus (family Iridoviridae), which comprises a group of emerging viruses that infect cold-blooded animals, including amphibians, fish, and reptiles
[1][2]. FV3 infections were first reported in leopard frogs in the 1960s, and several virus isolates were obtained from cultured tissues/cells of both healthy frogs and tumor-bearing ones with renal carcinoma
[1][2][3]. This implied tumorigenic potential; however, further studies demonstrated no etiological association of FV3 with the renal oncogenesis
[1][2][3]. On the other hand, the association of FV3 with apparently healthy frogs indicates host-adaptive transmission and persistence, and may de facto cause diseases in other susceptible stages during the amphibian life cycle
[4]. More studies have implicated Ranaviruses in the decline of amphibian populations worldwide
[5][6][7][8]. FV3 represents the most frequently reported iridovirus for anurans. In North America, FV3 is widespread in wild amphibians, and is the only Ranavirus detected in turtles
[6][9][10]. A recent study detected different FV3 lineages in wild amphibians in Canada, and these new FV3 isolates appears to have undergone genetic recombination with the common midwife toad virus (CMTV)
[9]. CMTV represents another Ranavirus to affect various amphibians and reptile species and cause mortality events throughout Europe and Asia
[9][10]. These findings reinforce the urgency to study ranaviral biology to face the bio-ecological threat from current catastrophic amphibian decline and negative impacts in aquaculture
[1][2][3][4][5][6][7][8][9][10].
Among various Ranaviruses accounting for epizootics in amphibians, fish, and reptiles, FV3 is the best-characterized model and the prototype of the genus Ranavirus
[1][2]. Historically, FV3 studies have provided insights into Ranavirus biology, including relevant characterization of highly methylated and phage-like genetic DNA, two-stage viral genome replication, temporal transcription, and virus-mediated arrest of the host response
[2][11]. Prompted by early studies of FV3’s DNA synthesis occurring at the two-stage fashion between the nucleus and cytoplasm
[11], more comparative genomic studies of large nuclear and cytoplasmic DNA viruses (NCLDVs) of eukaryotes have revealed the monophyletic origin of four viral families: poxviruses, asfarviruses, iridoviruses, and phycodnaviruses
[12][13][14]. As recent proposals extend NCLDVs to include three other taxonomic families (Ascoviridae, Marseilleviridae, and Mimiviridae) and new founding members of other types of giant dsDNA viruses, advances in Ranavirus research contribute to delineate viral evolution and host tropism diversity among iridoviruses and NCLDVs, which can unveil evolutionary links among viruses associated with different cellular life forms
[13][15]. However, in spite of the general characterization of FV3 replication and infection, the transcriptomic profile of the many viral genes and the precise roles of most viral proteins of FV3 and most other Ranaviruses remain elusive.
Early pioneering studies have resolved 47 viral RNAs and 35 viral proteins using gel electrophoresis in FV3-infected fish cells, and temporally classified them into early, immediate delay, and late genes along the viral infection cycle
[16][17]. The first report of transcriptomic analysis used both microarray hybridization and RT-PCR validation to examine the expression of all 98 coding genes (or open reading frames, ORFs) as annotated in the FV3’s reference genome
[18]. In that study, Majji et al. identified 33 immediate early (IE) genes, 22 delayed early (DE) genes, and 36 late (L) viral genes, while seven remaining genes were undetermined. As was postulated for the temporal class of FV3’s genes in general, early genes (including both IE and DE) encode putative regulatory factors, or proteins that act in nucleic acid metabolism and immune regulation, whereas products of L genes are involved in the virion packaging, assembly, and cellular interaction for viral release
[18]. Notably, all of these previous FV3 gene transcription studies were performed in vitro using a fathead minnow (FHM) fish cell line model
[16][17][18]. Thus, to date, FV3 transcriptomic profiling in vivo in infected host is lacking.
To complement recent virome studies and novel Ranavirus isolations, it is imperative to characterize de novo FV3’s transcriptome and conduct gene functional analysis using next generation sequencing (NGS)-facilitated metagenomics approaches
[19][20]. To establish a procedure for unbiased analyses of ranaviral gene expression on a genome scale, we have performed a whole transcriptomic analysis (RNA-Seq) using total RNA samples containing both the viral and cellular transcripts from FV3-infected frog tissues. Two FV3 strains, a wild type (FV3-WT) and an ORF64R-knockout strain (FV3-∆64R), were used for comparison
[21][22]. The gene 64R encodes a caspase-like activation and recruitment domain decoy (vCARD)-like molecule postulated to serve as an immune evasion gene. Recombinant FV3-∆64R virus exhibits attenuated virulence and growth in vivo, and exhibits a different host-pathogen interaction compared to wild-type FV3
[21][22]. In accordance with previous studies showing that FV3 replicates in multiple amphibian tissues
[16][17], our virus-targeted transcriptomic analysis specifically mapped reads spanning the full-genome coverage at ~10× depth on both positive and negative strands in samples from the infected intestine, liver, spleen, lung, and especially kidney. In contrast, reads were only mapped to fragmental regions in samples from the infected thymus, skin, and muscle. Importantly, no viral transcript reads were detected in all control mock-infected tissue samples, indicating a well-controlled experimental handling and contamination-free processing. Our analyses identified the expression of most of the 98 annotated ORFs and profiled their differential expression in a tissue-, virus-, and temporal class-dependent manner. Furthermore, we used a reverse-genetic approach to functionally identify viral putative ORFs that encode hypothetical proteins, particularly those containing viral mimicking domains analogical to that in host interferon (IFN) regulatory factors (IRFs) or IFN receptors, especially for the type III IFNs. As a cardinal antiviral mechanism diversified along tetrapod evolution, the IFN system comprises three types of IFNs (type I, II, and III) that are classified mainly based on their molecular signatures and type-specific cognate receptors
[23][24][25]. IFNs induce diverse immune responses extensively characterized in antiviral responses, and are involved in immunomodulatory processes through signaling cascades via respective IFN receptors and various IRFs
[23][24][25][26]. Previous studies have determined the key position of amphibians in IFN evolution
[24][25], and the alteration of viral infection in the IFN response in FV3-infected frogs
[21][22]. The functional analyses may provide a mechanistic explanation about the viral interference of IFN responses in FV3-infected cells/tissues, and provide new insights into the evolutionary arms race between the Ranavirus and quickly evolving amphibian IFN system
[21][22][23][24][25]. Our study thus provides the first virus-targeted, genome-wide transcriptome profiling of a large DNA virus during real amphibian host infection and uncovers the potential function of hypothetical proteins in the context of the virus-host interaction.
2. FV3 Infection and Comparative Viral Determination Between FV3-WT and FV3-∆64R Strains in the Kidneys of Adult Frogs
FV3 infects anuran amphibians at various developmental stages, and is highly lethal in tadpoles. Adult frogs, by contrast, are more resistant to viral infection, and after viral clearance, a low level of quiescent viruses were isolated from apparently healthy frogs
[21][22][27]. This indicated that adult frogs are more adaptive to the deathliness caused by the virus
[22], and meanwhile serve as active carriers or reservoirs for the virus transmission, providing a valuable model for studying the virus–host coevolution
[26]. In this context, we infected in laboratory-controlled conditions 1–2-year-old X. laevis frogs for well-controlled sample collection. As shown in
Figure S1 and
Figure 1, randomly allotted frogs were infected with either a FV3-WT or FV3-∆64R strain. Time points were chosen based on a previously published study to include an early innate immune response (1 dpi), intermediate response (3 dpi), and the peak of the adaptive T cell immune response (6 dpi)
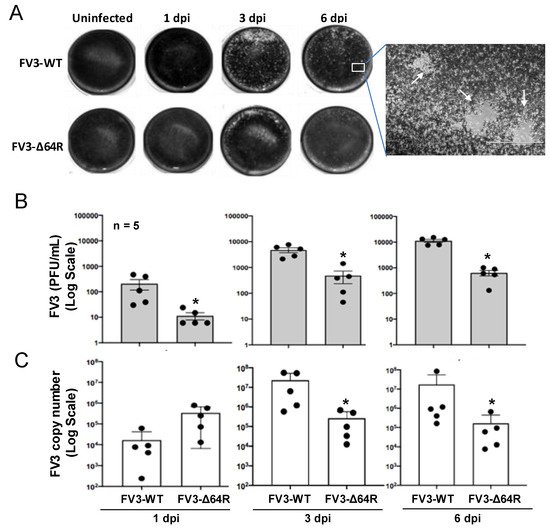
[3][4]. Both FV3 strains caused early productive infections, as shown with successful viral isolation from the kidney tissue homogenates of infected frogs (
Figure 1A). As anticipated, FV3-WT was more efficient in producing infectious virions compared to the FV3-∆64R mutant virus (with disruption of the ORF64R gene, encoding a putative interleukin-1 beta convertase and containing a caspase activation and recruitment domain (vCARD)), showing a 100–1000 magnitude difference compared to using a logarithmic scale of PFU at 1–6 dpi (
Figure 1B). Similar infection patterns were also observed by measuring the virus genome copy number based on quantitative detection of the viral Orf60R gene, which encodes a virus DNA polymerase II unit (Pol II) (
Figure 1C). However, the viral genome copy number of FV3-WT kept increasing through 1–6 dpi, whereas FV3-∆64R genome copies reached a higher level than the wild type at 1 dpi and kept a similar level through the tested period without much increase (
Figure 1C). From daily observations throughout the infection process, infected frogs behaved similarly to the mock-infected controls, and no outliers per clinical observation or virus diagnosis were identified, which were averagely qualified for sample collection, as designed for further transcriptomic processing (
Figure S1).
Figure 1. Viral plaque assays and genome copy number detection by quantitative PCR (QPCR). (A) For plaque assays, 1 mL of the virus-containing supernatant of tissue homogenate from an individual kidney sample was used to inoculate A6 cells (ATCC® CCL-102™). FV3 plaques were counted at 1, 3, and 6 days post-infection (dpi) and imaged for representative wells. (B) Virus titers were calculated to present as PFU/mL average for each group/treatment. (C) Virus genome copies were examined in the DNA samples from the infected kidneys using a routine QPCR procedure to determine the FV3gorf60R gene copies in 150 ng DNA per reaction as described. * p < 0.05, n = 5 for (B,C).
3. Virus-Targeting Transcriptome Analysis and Difference Dependent on Tissue Types and FV3 Strains
FV3’s genome encodes 98 putative coding genes (FV3gorf1-98) as annotated along its reference genome
[18]. Previous microarray analysis plus RT-PCR validation determined the expression of all 98 FV3 ORFs, indicating full-genome transcribing capacity during FV3 infection in the FHM cell line
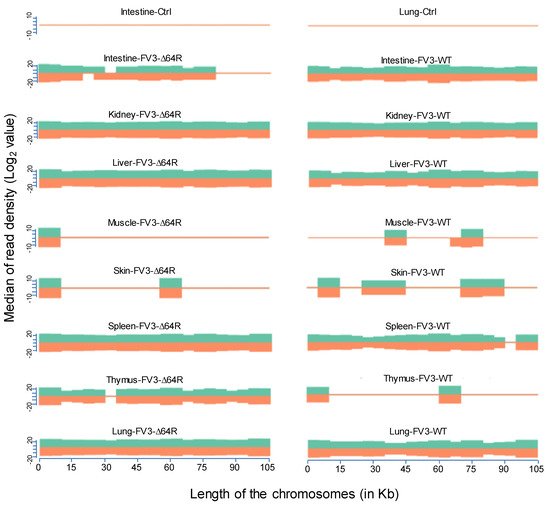
[18]. Consistent with the microarray analysis, our comprehensive unbiased transcriptome analysis based on de novo deep sequencing revealed FV3 gene-specific reads spanning the full FV3 genome at ~10× depth in samples from the infected intestine (FV3-WT only), liver, spleen (FV3-∆64R only), lung, and particularly kidney (
Figure 2). In addition, partial genome coverage or regional detection were obtained in FV3-infected muscle, skin, and thymus tissues (
Figure 2). Importantly, no FV3-specific reads were detected from any sham-infected control tissues, ruling out cross-contamination and validating our sample handling procedures.
Figure 2. Virus-targeted transcriptome analysis in the control and infected samples at 3 dpi. Shown is the distribution plots of mapped reads in the FV3 genome (GenBank accession no. NC_005946.1). The x-axis shows the length of the genome (in Kb, 105 Kb of FV3), and the y-axis indicates the log2 of the median of the read density. Green and red indicate the positive and negative strands, respectively. Note, no FV3 transcript reads were obtained from the control (Ctrl) non-infected samples (shown only from the intestine and lung).
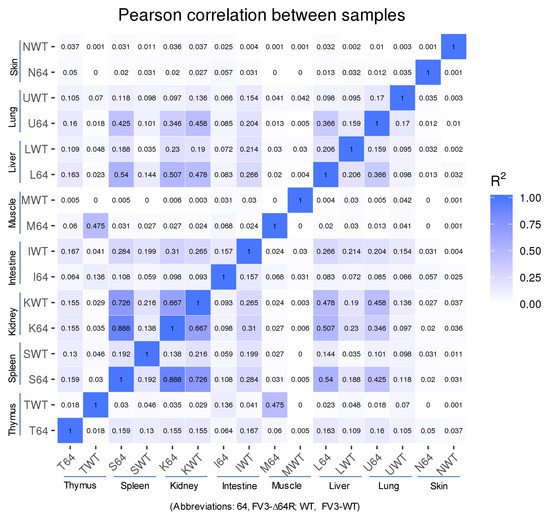
Statistical analyses of the RNA-Seq results revealed several interesting aspects: (1) FV3 may maintain a more complex transcript mixture in vivo than in a uniform cell line related to unsynchronized infection stages upon diverse cell types in tissues. Given that the viral transcripts were significantly detected in the kidney, spleen, liver, lung, thymus, and intestine, we concluded that the differential viral gene expression in different tissues resulted primarily from a systemic virus–host interaction initiated from intraperitoneal injection. (2) FV3-specific reads were significantly enriched in an increasing order in the intestine, lung, liver, spleen, and predominantly in the kidneys, but much less abundant in the skin and muscle, where FV3 replication may be negligible. (3) The transcript profiles of the FV3-∆64R mutant were nearly identical to the FV3-WT in the kidney, but qualitatively and quantitatively very different in other tissues. (4) The unbiased transcriptome study detected transcripts of the FV3 genome almost equivalently along both positive and negative strands, which confirmed the existence of viral coding genes at both strand orientations (Figure 2). Further co-expression Venn analysis confirmed a virus strain (FV3)-dependent difference of gene transcription among most tested tissues, as both WT and FV3-∆64R shared near identical transcript profiles of all 98 annotated FV3gorfs in the kidney. Interestingly, although WT FV3 genes were more efficiently transcribed than FV3-∆64R in the intestine, skin, and kidney, the recombinant mutant virus actually had much more transcripts in the thymus, liver, lung, and particularly spleen. This implies that the disruption of the FV3gorf64R gene, which encodes a putative interleukin-1 beta convertase containing caspase recruitment domain (vCARD), may change viral transcription dynamics and the tissue/cell tropism of FV3 infection in amphibians (Figure 2). As shown in Figure 3, the Pearson correlation analysis demonstrated a generally low cross-sample correlation, except between the FV3-∆64R-infected spleen and kidney, further indicating that both tissue types had a top priority to support FV3 infection and full-scale gene expression (Figure 3).
Figure 3. RNA-Seq correlation among the samples. Heat maps of the correlation coefficient between samples are shown. Numbers indicate the square of the Pearson coefficient (R2). The closer the correlation coefficient was to 1, the greater the similarity of the samples. Generally low R2 values indicated a dramatic difference between different tissues and two FV3 strains.
4. Conclusive Highlights
Frog virus 3 (FV3) represents a well-characterized model to study Ranavirus pathogens that are prevalent in worldwide habitats of amphibians, fish, and reptiles, and significantly contribute to the catastrophic amphibian decline
[5][6][7][8][9][10]. Based on conventional and novel assignation of FV3 coding genes per their temporal expression fashion along the virus infection stages
[16][17][18], the current study used an unbiased transcriptomic RNA-Seq analysis to profile and compare viral transcripts in various tissues of frogs infected with either FV3-WT or a FV3-∆64R strain defective for a gene encoding a CARD motive. The results revealed a full-genome coverage transcriptome annotated to almost all coding genes at ~10× depth on both positive and negative strands in RNA samples from the infected intestine, liver, spleen, lung, and especially kidney. In contrast, partial transcript coverage was detected in infected thymus, skin, and muscle tissue, suggesting inefficient viral replication in these tissues. Extensive analyses indicated a multi-organ infection pattern of FV3 infection in frogs and validated the in vivo expression of most annotated 98 ORFs, as well as their differential expression in a tissue-, virus strain-, and temporal class-dependent manner. About half of FV3’s coding genes have not yet been functionally determined in the scenario of the virus–host interaction. Our transcriptome-initiated functional analyses focused on putative ORFs that encode hypothetical proteins containing viral mimicking domains, such as host interferon (IFN) regulatory factors (IRFs) and IFN receptors. Our findings suggest that Ranaviruses like FV3 have acquired during evolution previously unknown molecular mimics interfering with host IFN signaling, which thus provide a mechanistic understanding about Ranavirus persistence in adult frogs. In summary, this study provides a comprehensive virus-targeted transcriptome analysis to profile the genome-wide gene expression of a large double-strand DNA virus, and uncovers the potential IFN-interfering function obtained by some ranaviral hypothetical proteins to perturb the virus–host interaction.
 +1 point
+1 point