2.1. Effect of Wnt/β-Catenin Signaling Pathway on Skeletal Fluorosis
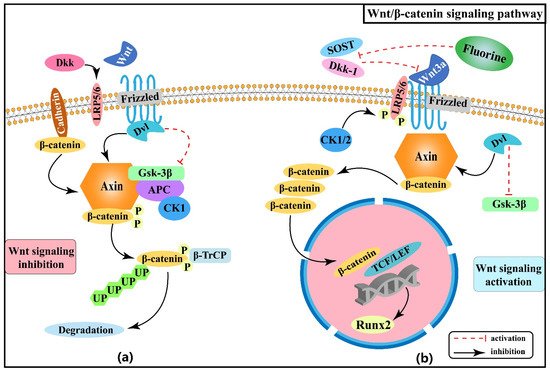
Wnt is a cytokine involved in various biological processes
[11], and the canonical Wnt/β-catenin signaling pathway plays a crucial role in regulating osteoblast differentiation, osteogenic matrix formation, and bone homeostasis
[12][13][14][15][16]. In the absence of Wnt ligands, however, the potential transcriptional mechanisms of the pathway cannot be activated
[17]. The β-catenin inhibiting action of glycogen synthase kinase-3β (GSK-3β) is hindered when Wnt ligands (Wnt1, Wnt2, Wnt3a, and Wnt10a) act on the cell surface receptors Frizzled and low-density lipoprotein receptor-related protein 5/6 (LRP5/6), leading to decreased phosphorylation and degradation of β-catenin. β-catenin is thereby able to accumulate to a high enough level to enter the nucleus and interact with T-cell factor/lymphatic enhancer factor (TCF/LEF), thus regulating the expression of the target gene
[17].
Sun et al. confirmed that fluoride significantly increased osteoblast activity, as evidenced by the observed enhanced osteogenesis in their study
[18]. Fluoride increased the expression of Wnt3a and β-catenin in rat osteoblasts; in addition, serum bone alkaline phosphatase (BALP) levels tended to increase with increasing doses of fluoride staining
[19], suggesting that fluoride may be involved in the bone formation process by stimulating Wnt3a to up-regulate BALP expression. A recent study also found that fluoride can induce abnormal activation of the Wnt/β-catenin signaling pathway, causing the increased formation of cancerous bone in mice and protein expression of Wnt3a and phospho-Gsk-3β, as well as its downstream target gene Runt-related transcription factor 2 (Runx2). However, it must be noted that inhibition of β-catenin can suppress fluoride-induced Runx2 protein expression and resulting osteopathology
[15], which suggests that β-catenin may be a key molecule in fluoride-induced aberrant osteogenesis. Serum sclerosing protein (SOST) and dickkopf-related protein 1 (Dkk-1) are inhibitors of Wnt/β-catenin signaling, and Wang
[20], Liu
[21], and Zeng et al.
[22] have demonstrated that long-term or high-concentration exposure to fluoride can reduce SOST and Dkk-1 concentration, thereby inducing the activation of Wnt/β-catenin signaling, and finally leading to the progression and differentiation of osteoblasts.
Figure 1 shows the association between Wnt/β-catenin signaling pathway and the pathogenesis of skeletal fluorosis.
Figure 1. Wnt/β-catenin signaling pathway in the pathogenesis of skeletal fluorosis. (a) Wnt signaling is inhibited; (b) Wnt signaling is activated.
Here, we review the mechanism of action of the Wnt/β-catenin signaling pathway in skeletal fluorosis, and it is evident that this signaling mediates the enhanced osteogenic effects of fluoride. Moreover, Wnt/β-catenin signaling pathway is connected to many signaling pathways in the control of osteoblast and chondrocyte proliferation and differentiation, such as PIK/Akt and hedgehog (Hh) signaling, and subsequent studies to explore their interactive regulation are even more valuable for comprehension of the pathogenesis of skeletal fluorosis.
2.2. Effect of Notch Signaling Pathway on Skeletal Fluorosis
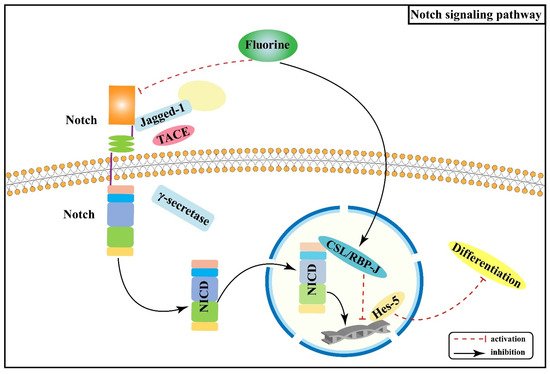
Notch signaling pathway primarily mediates intercellular interactions and plays an integral part in determining cell fate and function together with the regulation of skeletal homeostasis
[23]. The interactions of Notch receptors (Notch-1, 2, 3, and 4) with ligands (Jagged-1, Jagged-2, and Delta-like-1, 3, and 4) lead to a series of proteolytic cleavage and release of the Notch intracellular structural domain (NICD) into the cytoplasm
[24][25]; NICD translocates to the nucleus and forms a complex with Epstein-Barr virus latency C promoter binding factor 1 (CBF-1)/repressor of hairless/Lag1 (CSL), which is known as recombination signal binding protein-Jκ (RBP-J) in mice, and Mastermind-like (Maml) to activate transcription of target genes (Hes-1, Hes-5, Hey-2, and Hey L)
[26][27][28]. Notch signaling pathway regulates the development of osteoblast and osteoclast lineages and thus has a significant impact on skeletal development
[29].
Notch signaling can inhibit osteoblast differentiation
[30][31], thereby maintaining bone marrow mesenchymal progenitors
[27]. An animal experiment found that excessive fluoride exposure decreased the protein as well as mRNA expression levels of Notch-3 and Jagged-1 in rats, especially in osteoblasts
[32], suggesting that fluoride can inhibit the Notch signaling pathway, thus promoting osteoblasts proliferation and differentiation, disturbed dynamic homeostasis of bone tissue, and the pathological manifestation of osteosclerosis. Another study
[33] showed that RBP-J protein expression was increased and Hes-5 protein expression was decreased in bone tissue of rats dyed with fluoride, indicating that fluoride may promote osteoblast differentiation by promoting the expression of the transcriptional repressor RBP-J and enhancing the inhibitory effect on the downstream target gene Hes-5. Prior studies have suggested that the Notch signaling pathway mediates the enhanced osteogenic effects of fluorosis.
Figure 2 shows the association between Wnt/Notch signaling pathway and the pathogenesis of skeletal fluorosis.
Figure 2. Notch signaling pathway in the pathogenesis of skeletal fluorosis.
Notch signaling pathway displays a significant role in cell proliferation, differentiation, and apoptosis. Nonetheless, there are relatively few investigations on the relationship between Notch signaling pathway and skeletal fluorosis, mainly focusing on the mechanisms that regulate the proliferation and differentiation of osteoblasts. An in-depth investigation of the role of Notch signaling pathway in regulating other bone tissues and cells and its synergistic or antagonistic effects with other signaling pathways is of great significance for the pathogenesis, prevention, and treatment of skeletal fluorosis.
2.3. Effect of PI3K/Akt/mTOR Signaling Pathway on Skeletal Fluorosis
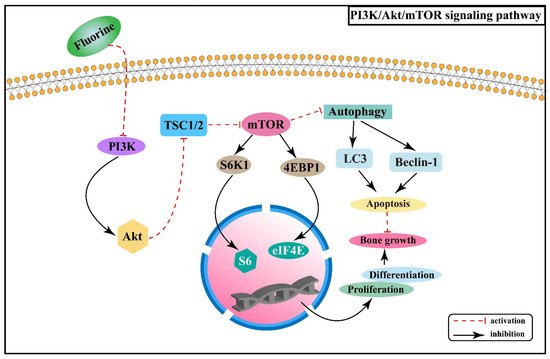
mTOR is a serine/threonine-protein kinase that can regulate a variety of biological processes. PI3K/Akt/mTOR signaling pathway is involved in the proliferation and differentiation of osteoblasts, osteoclasts, and chondrocytes
[34][35][36]. Experiments in rats with chronic fluorosis have shown that the PI3K/Akt signaling may cause excessive proliferation and differentiation of osteoblasts and accelerate bone turnover, resulting in osteosclerotic skeletal fluorosis
[37]. It was found that high-dose fluorine can inhibit the expression of mTOR protein in cartilage tissue, promote the expression of autophagy signature proteins, such as Beclin-1 and cytoplasm-associated protein light chain 3 (LC3), and enhance the apoptosis involved in the process of fluorine-induced cartilage damage
[38]. After mTOR is activated, it phosphorylates the downstream target proteins ribosomal protein S6 kinase β-1 (S6K1) and eukaryotic translation initiation factor 4E binding protein 1 (4EBP1) to promote gene transcription and protein translation
[39]. Fluorine can down-regulate PI3K/Akt/mTOR signaling and inhibit the phosphorylation of its downstream factors S6K1 and 4EBP1, thus promoting chondrocyte autophagy and inhibiting chondrocyte proliferation and differentiation
[40]. The association between PI3K/Akt/mTOR signaling pathway and the pathogenesis of skeletal fluorosis is shown in
Figure 3.
Figure 3. PI3K/Akt/mTOR signaling pathway in the pathogenesis of skeletal fluorosis.
Skeletal fluorosis is characterized by a disrupted dynamic balance between bone resorption and formation and is intimately linked to regulating PI3K/Akt/mTOR signaling pathway, with a critical role in osteoblast proliferation and differentiation as well as chondrocyte autophagy. Understanding more about the changes of PI3K/Akt/mTOR signaling pathway and related signaling molecules in skeletal fluorosis, as well as its interaction with other signaling pathways, will promote a profound understanding of the pathogenesis of endemic fluorosis and provide a basis for the prevention and treatment of skeletal fluorosis.
2.4. Effect of Hedgehog Signaling Pathway on Skeletal Fluorosis
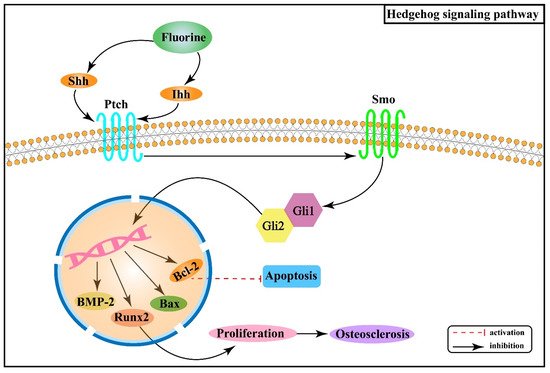
Hh signaling pathway consists of Hh protein, protein receptors Ptched (Ptch) and Smoothened (Smo), and the 5-zinc finger transcription factor Glis (Gli1, Gli2, and Gli3)
[41]. There are three Hh homolog genes in mammals: Sonic Hedgehog (Shh), Indian Hedgehog (Ihh), and Desert Hedgehog (Dhh), which encode Shh, Ihh, and Dhh proteins, respectively. In the absence of Hh ligands, Ptch inhibits the activity of Smo, which further inhibits the expression of downstream genes in the nucleus
[42]. When the Hh ligand binds to Ptch, its inhibitory effect on Smo is released, and the expression of target genes increases
[42]. Research results have shown that removing Smo decreased the proliferation rate of chondrocytes, while upregulation could increase the rate
[43].
Studies have revealed that fluoride stimulation increases the expression of Ihh in rat osteoblasts, and this signal, mediated by Smo, induces the expression of Runx2 after activating Gli2, which in turn promotes osteoblast proliferation and contributes to osteosclerosis
[44]. Moreover, fluorine stimulation can cause chondrocytes to produce oxygen radicals, thus causing abnormal differentiation
[45][46]. Shh is stimulated and released when oxidative stress occurs, synergizing with oxidative stress to promote the B cell lymphoma/leukemia-2 (Bcl-2) anti-apoptotic protein
[47]. Animal experiments demonstrated that with the expression of Shh, Smo, bone morphogenetic protein-2 (BMP-2) and B cell lymphoma/leukemia gene associated × (Bax) protein gradually increased in rat cartilage tissues with increasing fluoride staining concentration, while Bcl-2 protein expression gradually decreased
[48].
Figure 4 shows the association between Hh signaling pathway and the pathogenesis of skeletal fluorosis.
Figure 4. Hedgehog signaling pathway in the pathogenesis of skeletal fluorosis.
The above studies suggest that the Hh signaling pathway is closely associated with oxidative stress and apoptosis and regulates chondrocyte ossification and apoptosis in skeletal fluorosis. In addition, the available data reveal that Hh signaling is also related to Wnt/β-catenin, Notch, Bmp, and other signaling pathways, and its specific regulatory mechanisms in the pathogenesis of skeletal fluorosis that damages cartilage tissue remain to be investigated in depth.
2.5. Effect of Hormones and Their Receptor Signaling Pathways on Skeletal Fluorosis
Over the years, systematic studies of the mechanisms of fluorosis in animals and humans have revealed that many calcium-regulating hormones and cytokines are also involved in mediating abnormal osteogenic and osteolytic bone metabolism in skeletal fluorosis, such as parathyroid hormone (PTH) and transforming growth factor β (TGF-β). PTH is a polypeptide hormone responsible for regulating calcium homeostasis in the body and exerts an active role in regulating bone turnover. Its effects on cells are mediated by the G protein-coupled receptor (PTH receptor, PTH1R) expressed by target cells, including osteoblasts, osteoblast precursor cells, and osteocytes, but excluding osteoclasts
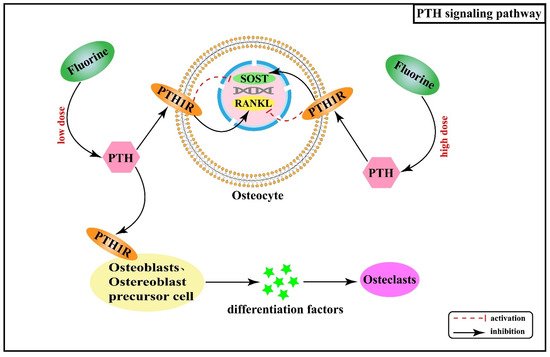
[49][50]. Therefore, PTH acts indirectly on osteoclasts by binding to PTH1R on the surface of the osteoblast cell line and secreting osteoclast differentiation factors. Additionally, a study found that serum PTH levels gradually increased with increasing fluoride exposure
[51]. PTH can regulate fluoride action through its effects on the expression of SOST and receptor activator of nuclear factor-κB ligand (RANKL) in osteocytes. By up-regulating RANKL and inhibiting SOST, PTH enhances the effectiveness of low-dose of fluoride in bone turnover promotion while having opposite effects on SOST and RANKL in the cases of high fluoride doses
[52].
Figure 5 shows the association between PTH signaling pathway and the pathogenesis of skeletal fluorosis.
Figure 5. PTH signaling pathway in the pathogenesis of skeletal fluorosis.
In addition, PTH, as one of the important hormones regulating calcium and phosphorus metabolism in the body, exerts a bidirectional regulatory effect on bone metabolism. High doses of PTH promote bone resorption, while at low doses, it stimulates bone formation. Furthermore, animal and clinical studies have revealed that when PTH is given intermittently subcutaneously, it stimulates bone formation, increases bone density in long bones and vertebrae, and improves the bone quality
[53][54]. It follows that the use of PTH in the treatment of osteoporosis has been approved by the US Food and Drug Administration and other organizations
[54].
Insulin is a multifunctional protein hormone that acts by binding to the insulin receptor (IR) in most tissues. IR and intracellular signaling pathways are the main components of the insulin signaling pathway
[55]. Fulzele et al.
[56] found that IR exists in osteoblasts, and insulin promotes bone formation by inhibiting Twist2 (Runx2 inhibitor) after interacting with cell surface receptors. Studies have pointed out that fluoride can stimulate IR expression and osteoblast function in vitro and affect insulin secretion, activity, and sensitivity
[57][58]. Furthermore, insulin state in turn interferes with bone formation and absorption
[57][58]. Another study showed that insulin plays a vital role by mediating IR signals, and insulin-like growth factor-1 (IGF-1) may play a role in bone turnover induced by excessive fluoride by regulating IR or its downstream
[59]. All these studies suggest that insulin exerts an essential role in fluoride-induced bone pathogenesis.
2.6. Interactive Regulatory Networks among Signaling Pathways Involved in Skeletal Fluorosis
In the pathogenetic progression of skeletal fluorosis, both local signaling pathways and hormones are involved. Activation or inhibition of signaling is not accomplished linearly within the single signaling pathway mentioned above alone, but rather the signaling pathways interact to form an interactive regulatory network that acts to control biological processes in a synergistic or antagonistic manner
[60].
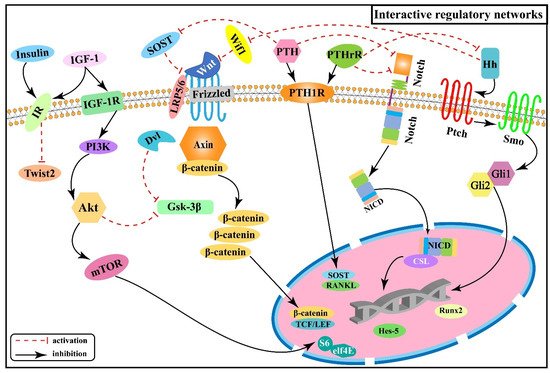
Figure 6 illustrates the interactive regulatory networks formed among Wnt/β-catenin, Notch, PI3K/Akt/mTOR, Hh, PTH, and insulin signaling pathways involved in skeletal fluorosis.
Figure 6. Interactive regulatory networks among Wnt/β-catenin, Notch, PI3K/Akt/mTOR, Hh, PTH, and insulin signaling pathways involved in skeletal fluorosis.
PTH can bind to the same receptor (PTH1P) as the parathyroid hormone-related peptide (PTHrP) synthesized by osteoblasts and mediates bone formation and accelerated bone turnover through multiple pathways. And SOST expressed by osteoblasts is a key negative regulator of bone formation
[61][62] and acts as an upstream inhibitor of the Wnt/β-catenin signaling pathway by binding to Wnt and blocking its interaction with the cell surface receptors Frizzled or LRP5/6 to inhibit the activation of β-catenin downstream in the Wnt signaling pathway. Whereas PTH causes transcriptional suppression of the osteocyte marker gene SOST and downregulates SOST protein expression
[63], thereby activating the Wnt/β-catenin signaling pathway. Experiments revealed that the expression of Ihh in rat growth plate chondrocytes decreased with increasing fluorine concentration, while the expression of PTHrP showed an increasing trend, suggesting that fluorine reduces the expression of Ihh by up-regulating PTHrP, inhibiting the Ihh/PTHrP negative feedback loop to affect chondrocyte proliferation and differentiation, and inhibiting the normal process of osteogenesis within cartilage
[64]. In addition, it was shown that PTH can down-regulate the expression of the Notch signaling pathway, and this inhibitory effect may be achieved by down-regulating intracellular cAMP/PKA signaling, reducing the expression of receptor Notch-1 and ligand Jagged-1 of the Notch signaling pathway, which in turn affects the expression of Runx2
[65]. The researchers found positive regulation among PTHrP, NICD, and Jagged-1 proteins after transfecting epiphyseal stem cells with PTHrP over-expression and lentiviral interference vectors, suggesting that PTHrP may act through influencing the Notch signaling pathway
[66]. The above information indicates that the Wnt/β-catenin, Hh, and Notch signaling pathways are associated with the PTH signaling pathway and act downstream of the PTH signaling.
Both Wnt/β-catenin and Ihh signaling pathways control the proliferation and differentiation of osteoblasts and chondrocytes at multiple stages
[12][13][14][15][44][45][46]. One study tested the genetic relationship between Wnt and Hh signaling by generating double mutant mice with results revealing that Wnt/β-catenin signaling acts downstream of Hh signaling in enhancing bone formation
[67], in agreement with another study
[68]. Subsequently, the expression of genes associated with different stages of osteoblast differentiation was also examined, showing that β-catenin is required downstream of Ihh in promoting osteoblast maturation
[67]. It has been demonstrated that Wif1 (Wnt inhibitory factor 1) exerts biological effects by mediating and regulating Shh/Wnt/β-catenin signaling
[69], as well as Wif1 can effectively block the activation of the canonical Wnt signaling pathway in chondrocytes by binding to Wnt ligands (Wnt3a, etc.)
[69][70][71], speculating Hh signaling may exert inhibitory effects on downstream Wnt/β-catenin signaling through Wif1 in the pathogenesis of skeletal fluorosis. At the same time, the Wnt/β-catenin signaling is also interactively regulated with the PI3K/Akt signaling. It was shown that PI3K/Akt negatively regulates Gsk-3β activity, thereby inhibiting the phosphorylation of β-catenin
[72], which suggests a role for Wnt/β-catenin signaling acting downstream of PI3K/Akt. To determine this role, the researchers further analyzed the expression of Wnt-regulated genes, such as Dkk1 and Sfrp1 (secreted frizzled-related protein 1), showing that PI3K signaling activates these genes by peptide-mediated α5β1 integrin priming in mesenchymal skeletal cells
[73]. Moreover, PI3K/Akt signaling is also regulated by insulin-related signaling. IGF-1 is a principal growth-promoting signal for vertebrate skeletal development
[74] and as a specific ligand can activate the PI3K/Akt signaling pathway by binding and phosphorylating the membrane IGF-1 receptor (IGF-1R), leading to osteoblast differentiation and proliferation
[75].
At present, studies on the relevant signaling pathways involved in skeletal fluorosis are still predominantly single lineage studies. Nevertheless, active osteogenesis and accelerated bone turnover as crucial processes in the progression of skeletal fluorosis are regulated by sophisticated networks of multiple signaling pathways. The profound exploration of the interactive regulatory mechanisms among signaling pathways in skeletal fluorosis will facilitate the development of specific targeted therapeutic measures.
 +1 point
+1 point