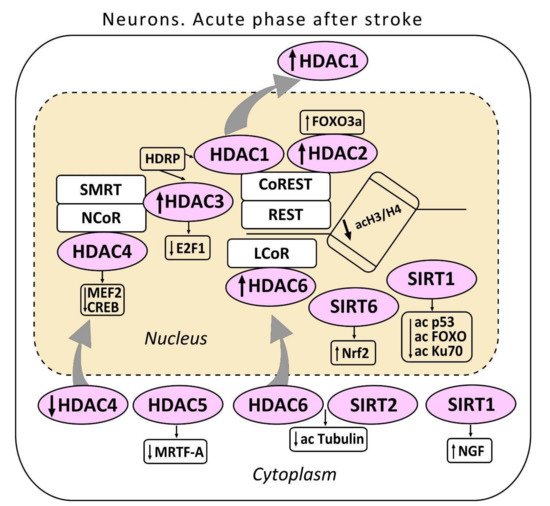
3.1. Class I HDACs
The first class HDACs is widely represented in the brain
[31]. HDAC1 localizes both in the neuronal nuclei, and in the cytoplasm, where it deacetylates some cytoplasmic proteins. HDAC2 localizes exclusively in the neuronal nuclei
[22][33]. HDAC1 suppresses the production of proteins, which regulate the cell cycle in somatic cells. It also contributes to cell protection against DNA damage
[34]. HDAC1 can serve as a molecular switch between neuronal survival and death
[35]. HDAC2 regulates apoptosis in the ischemic penumbra
[33][36]. HDAC1 and HDAC2 can be included in the multienzyme complexes Sin3, NuRD, CoREST, or NODE that suppress transcription of different sets of target genes
[37][38][39]. The complex CoREST suppresses genes involved in synaptic plasticity and post-stroke recovery
[40][41] (
Figure 1).
The upregulation of HDAC2, but not HDAC1, in the PTS-induced penumbra was associated with development of apoptosis
[22]. Other authors also showed the critical role of HDAC2 in death of neurons in the peri-infarction area after ischemic stroke. The upregulation of HDAC2 observed in the early recovery phase from five to seven post-stroke days reduced survival of neurons and augment neuroinflammation. HDAC2 targeting is apparently a novel therapeutic strategy for stroke recovery
[36]. MCAO-induced overexpression of HDAC2 decreased the number of synapses, impaired synaptic plasticity, reduced memory, and deteriorated other cerebral functions
[42][43]. Knockdown or knockout of the HDAC2 gene restored brain functions due to plasticity of the surviving neurons in the peri-infarction zone
[43][44].
The overexpression of HDAC1 and HDAC2 in ischemic penumbra neurons and white matter glial cells was observed in the mouse brain during the early regeneration period, 1 week after MCAO. Their levels in the infarct core, oppositely, decreased
[31]. Long-term overexpression of HDAC2 and HDAC8 was observed in neurons and astrocytes at 3‒14 days after photothrombotic stroke in the mouse cerebral cortex
[45]. Thus, HDAC2 deteriorates synaptic processes, impairs memory, disturbs various cerebral functions, and stimulates apoptosis in the ischemic brain.
HDAC3 also deacetylates histones H3 and H4 and some non-histone proteins. It also contributes to regulation of DNA replication and repair. The complex HDAC3/NCOR/SMRT is essential for maintaining chromatin structure and genome stability
[46]. Overexpression of HDAC3, HDAC6, and HDAC11 was observed in the ischemic penumbra 3 and 24 h after MCAO, and persisted for a week after reperfusion. The upregulation of HDAC3 and HDAC6 in the mouse cortical neurons was also observed in vitro in the neuronal cell culture. The inhibition of HDAC3 or HDAC6 expression by the short hairpin shRNA increased cell viability. This suggested their involvement in ischemia-induced neurotoxicity
[47]. The ischemia-induced neurotoxicity of HDAC3 was demonstrated in other studies
[35][48][49]. The neurotoxic effect of HDAC3 was associated with its binding to HDAC1. Actually, the knockdown of HDAC3 suppressed the neurotoxicity of HDAC1, whereas HDAC1 knockdown suppressed the neurotoxicity of HDAC3. HDAC3 and HDAC1 interact with histone deacetylase-related protein (HDRP), a shortened form of HDAC9, whose expression was reduced during neuronal death. The interaction between HDRP and HDAC1, but not HDAC3 protected neurons. HDRP inhibited the HDAC1/HDAC3 interaction and prevented the neurotoxic effect of any of these proteins. This is a possible mechanism of HDAC1-mediated action as a switch “survival/death” in cerebral neurons. HDAC1 interaction with HDRP promotes neuron survival, whereas its interaction with HDAC3 leads to apoptosis
[50]. On the other hand, HDAC3 was shown to suppress the production of the pro-apoptotic transcription factor E2F1 in neurons, and thus to contribute to survival of these cells
[51].
Another class I histone deacetylase HDAC8 is present mainly in the cytoplasm of neurons and astrocytes of the cerebral cortex, amygdala, hippocampus, and hypothalamus
[45][52]. The expression of HDAC8 in the mouse cortical neurons and astrocytes increased significantly during the recovery period, from 3 to 14 days after photothrombotic stroke
[43].
3.2. Class II HDACs
The data on the role of HDAC4 in neurodegeneration and neuroprotection are contradictory. On one hand, some authors have reported the ability of HDAC4 to maintain neuronal survival
[53][54][55]. However, other authors did not find a dependence of neuronal survival on HDAC4 expression
[56][57]. In cultured neurons, HDAC4 rapidly translocates into the nucleus under glutamate release, or decreased K
+ concentration in the medium. This stimulated cell death
[58][59]. The administration of brain-derived neurotrophic factor (BDNF) prevented nuclear translocation of HDAC4
[60]. On the contrary, inhibition or loss of calmodulin-dependent kinase IV (CaMKIV) stimulated HDAC4 accumulation in the neuronal nuclei
[59][61]. Nuclear HDAC4 was shown to promote neuronal apoptosis by suppressing the activity of prosurvival transcription factors MEF2 (myocyte enhancer factor 2) and CREB (cAMP response element-binding protein)
[58]. Other authors have reported that HDAC4 translocation into the nuclei of neurons, but not astrocytes, did not cause apoptosis in the MCAO-induced ischemic penumbra. Moreover, the nuclear localization of HDAC4 promoted post-stroke brain recovery
[62]. The HDAC4 level in the cytoplasmic, but not nuclear fraction of the rat brain cortex decreased at 24 h after photothrombotic stroke
[33]. The downregulation of HDAC4 and its relocalization into the neuronal nuclei continued during the recovery period, 2 weeks after stroke
[63][64]. In the neuronal nuclei HDAC4 deacetylates histones H3 and H4 and decreases the levels of some prosurvival proteins that finally lead to the neuronal death
[58][59][64]. Since HDAC4 was assumed to be inactive against histones, these effects could be mediated by its interaction with other nuclear proteins. Actually, HDAC4 was shown to exhibit deacetylase activity after interacting with the co-repressor complex HDAC3/NCOR
[65]. Further studies of HDAC4 interactions with different proteins are needed to understand its role in survival and death of cerebral cells after stroke.
HDAC5, another member of class II histone deacetylases, is involved in neuronal differentiation and axon regeneration in the injured sensory neurons
[66][67]. The overexpression of HDAC5 and its nuclear localization was shown to be associated with apoptosis of the cultured neurons from the cerebellar granular layer
[68]. After transient MCAO, HDAC5 suppressed the antiapoptotic effect of the transcription factor MRTF-A (myocardial transcription factor-A) in the rat brain neurons
[69]. HDAC5 expression in the ischemic penumbra decreased 1, 2, and 14 days after MCAO
[47][64]. The downregulation of HDAC5 in the mouse cerebral cortex was observed at 3 days after photothrombotic stroke. However, the number of the apoptotic HDAC5-positive cells did not change
[63]. Possibly, the decrease in the level of HDAC5 in cortical neurons was associated with the regeneration processes
[70].
HDAC6 belongs to the IIb class of histone deacetylases. It is involved in various cellular processes such as degradation of damaged proteins, cell migration, and intercellular interactions
[71]. One of the cytoplasmic substrates of HDAC6 is α-tubulin. Its deacetylation induced destabilization of microtubules in the course of cytoskeleton reorganization and axonal growth during post-stroke regeneration
[72]. In the mouse or rat brains HDAC6 presents not only in the cytoplasm, but also in the nuclei of some cortical neurons, but not astrocytes
[33][63]. During the first two weeks after the photothrombotic stroke, HDAC6 was upregulated in the neurons not only in the penumbra, but also in the contralateral cerebral cortex, where it appeared in the neuronal nuclei. In the PTS-induced penumbra, HDAC6 co-localized with apoptotic neurons that indicated its involvement in neuronal apoptosis
[57][63].
Thus, HDACs are widely represented in the brain. The expression of HDAC1, HDAC2, HDAC3, HDAC4, and HDAC6 increased in the ischemic penumbra. Some of them are located in the neuronal nuclei, some in the cytoplasm, and others—both in the nucleus and cytoplasm. Their functions after ischemic stroke differed. Some HDACs mediate prosurvival processes, whereas others are involved in neurotoxicity. HDAC2 and HDAC6 were apparently involved in apoptosis in the post-ischemic brain.
3.3. Sirtuins
Sirtuins (SIRT) are class III histone deacetylases. The coenzyme nicotinamide adenine dinucleotide (NAD
+) makes sirtuins sensitive to metabolic and redox changes
[73]. In mammals, seven sirtuins have been identified. Of these, SIRT1 and SIRT6 are localized mainly in the cell nuclei, SIRT7 in the nucleoli, SIRT2 in the cytoplasm, and SIRT3, SIRT4, and SIRT5 are mitochondrial proteins
[74]. Sirtuins deacetylate a variety of substrates such as transcription factors, enzymes, and histones. They control diverse biological processes including metabolism, cell growth, aging apoptosis, and autophagy
[75]. In the present review we focus on the role of non-mitochondrial SIRT1, SIRT2, and SIRT6 in the brain damage and recovery after ischemic stroke.
SIRT1 content in the brain is higher than in other organs
[74]. In the hippocampus it regulates synaptic plasticity and memory. Since SIRT1 deacetylates histones and various transcription factors
[72][76], and, also, has the chaperone-like activity
[77], its subcellular localization is of significant importance for its functioning. The presence of the nuclear localization signal (NLS) and the nuclear export signal (NES) in the SIRT1 molecule allows it to shuttle from the nucleus to the cytoplasm and back that was assumed to be required for synaptic plasticity and memory formation
[78][79]. The subcellular location of SIRT1 changed during brain development and in response to physiological and pathological stimuli
[79][80].
SIRT1 mediates neuroprotection after ischemic stroke, traumatic brain injury, and neurodegenerative diseases. It regulates neurogenesis, neurite outgrowth, and gliogenesis, which are involved in postischemic brain regeneration
[76][81]. In the SIRT1 knockout mice, MCAO induced greater cerebral infarction than in control animals
[82]. On the contrary, mice overexpressing SIRT1 were more resistant to ischemia than control animals
[83]. The activation of SIRT1 by resveratrol reduced the MCAO-induced infarction volume
[84]. SIRT1 was overexpressed in the ischemic penumbra 7 days after MCAO in the mouse cerebral cortex
[82]. Nuclear SIRT1 was reported to prevent apoptosis by deacetylation of proteins p53
[85], FOXO
[86], and Ku70
[87]. On the contrary, SIRT1 localized in the cytoplasm enhanced caspase-dependent cell apoptosis
[88]. Nevertheless, the translocation of SIRT1 into the cytoplasm was not associated with cell apoptosis in the peri-infarct area at 7 days after photothrombotic stroke in the mouse cerebral cortex. In this case, the cytoplasmic localization of SIRT1 was associated with the upregulation of synaptophysin and GAP-43 that mediate the axon outgrowth and restoration of synaptic connections
[89]. The cytoplasmic SIRT1 was shown to enhance the neurite outgrowth that was induced by nerve growth factor NGF. Oppositely, inhibitors of SIRT1 or SIRT1-siRNA significantly reduced this effect
[90].
SIRT2 is expressed predominantly in oligodendrocytes and in the myelin-rich regions of the ischemic brain. It was not found in astrocytes, microglia, or neurons
[91]. However, other authors reported the presence of SIRT2 in the cytoplasm of neurons, but not astrocytes in the mouse cerebral cortex
[89][92]. In the cytoplasm, SIRT2 such as HDAC6 regulates the microtubule dynamics through deacetylation of α-tubulin
[93]. SIRT2 and HDAC6 can deacetylate α-tubulin either together
[94], or separately
[93]. Interestingly, the inhibition of SIRT2 increased acetylation of the microtubular α-tubulin mainly in the perinuclear zone, whereas inhibition of HDAC6 caused the general hyperacetylation of microtubules throughout the cell
[95]. Although some studies pointed to the pathological role of SIRT2 after cerebral ischemia
[76][96], the functions of SIRT2 in the ischemic brain are possibly more complicated than only pathological or only neuroprotective. Indeed, transient MCAO reduced the expression of SIRT2 and its translocation into the neuronal nuclei that played a neuroprotective role
[96]. On the contrary, the overexpression of SIRT2 in the cytoplasm of the cerebellar neurons or in vitro in the differentiated PC12 cell line was shown to induce apoptosis
[77][97]. Sirt2 was shown to mediate the myelin-dependent neuronal dysfunction during the early phase after MCAO in the mouse brain. Notably, the dynamics of Sirt2 mRNA and the protein level after ischemia differed
[91].
SIRT6 was found in both: cerebral neurons and astrocytes
[89][98]. It deacetylates mainly the lysine residues 9 and 56 in histone H3 that possibly represses genes associated with aging
[99]. The role of SIRT6 in ischemia is still unclear and controversial. On one hand, SIRT6 protected the brain from postischemic reperfusion injury due to stimulation of transcription factor Nrf2 (nuclear factor-like (erythroid 2)-like 2), which regulates the expression of antioxidant proteins and suppresses oxidative stress
[100][101]. SIRT6 was co-expressed with GAP-43, a marker of axon growth and synapse formation, at 14 days after photothrombotic stroke in the mouse cerebral cortex
[89]. Whether SIRT6 functions as a part of the multiprotein complex in the postsynaptic membranes
[102], or it regulates the neurite growth during the post-stroke recovery phase should be further studied. SIRT6 immunofluorescence was not observed in apoptotic cells in the PTS-induced penumbra
[89]. On the other hand, its overexpression in cultured neurons under oxygen and glucose deprivation was associated with necrosis of cortical cells
[103].
Thus, sirtuins, SIRT1 and SIRT6, are involved in the postischemic brain regeneration. SIRT1 regulates synaptic plasticity, memory, neuritogenesis, neurogenesis, and gliogenesis. SIRT6 protects neurons and astrocytes from the post ischemic reperfusion injury via stimulation of the transcription factor that regulates the production of antioxidant proteins Nrf2.
The HDAC inhibitors that have been developed to date are capable of inhibiting almost all HDAC isoforms of these four classes with varying degrees of specificity.
 +1 point
+1 point