The role of extracellular vesicles (EVs) proteome in diffuse large B-cell lymphoma (DLBCL) pathology, subclassification, and patient screening is unexplored. Here, we used an in vitro model of Germinal center B-cell like (GCB - good prognosis) and activated B-cell (ABC - poor prognosis) subtypes to propose potential drug targets and biomarkers.
1. Introduction
B-cell lymphomas develop from B-lymphocytes and account for 85% of all non-Hodgkin lymphomas (NHLs). Diffuse large B-cell lymphoma (DLBCL) is the most common form of lymphoma, accounting for 25% to 30% of all newly diagnosed cases of NHL. DLBCL is also a heterogeneous entity, encompassing a number of morphologic variants, distinct biologic entities, and variable clinical behaviors and responses to treatment
[1].
DLBCL is an aggressive form of NHL, infiltrating organs other than the lymph nodes at presentation in about 40% of the cases and with suboptimal outcomes in a fraction of patients. Additionally, some DLBCL subtypes are defined by organ location and have particular biological characteristics. The disease demonstrates remarkable progression-free survival (PFS) and overall survival (OS) rates in clinically and biologically defined subgroups of patients
[2]. Unfortunately, approximately 40% relapse or develop refractory disease upon standard RCHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone) treatment. Gene expression profiling (GEP) is currently the gold standard for determining the cell of origin (COO). GEP of DLBCL revealed two main subtypes: GCB (germinal center B-cell like) and non-GCB. Non-GCB tumors include the unclassifiable and the activated B-cell like (ABC) DLBCL subtypes; the latter is associated with poor treatment outcomes
[2][3]. In addition, a rarer primary mediastinal B-cell type was also identified by GEP.
The current limitations of the gold-standard method for DLBCL molecular subtyping (frozen tissue Affymetrix-based method) are associated with RNA isolation. Furthermore, its susceptibility to degradation by chemical treatments such as those used in formalin-fixed paraffin-embedded (FFPE) tissue preparation raises challenges for standardization. Fresh tissue biopsies are in some cases unavailable. Thus, the development of practical and reliable diagnostic methods derived from liquid biopsies has the potential to ameliorate the limitations of current methods and additionally improve monitoring of minimal residual disease. Current diagnostics based on immunohistochemistry (IHC), which serves as surrogate for COO, are not accurate despite the many algorithms proposed
[4]; in addition, traditional gene expression profiling is not applicable to routine diagnosis. Newer approaches such as nanostring technology are not generally used due to cost and the need for good quality tissue RNA. In order for targeted therapies to succeed in DLBCL, minimal invasive robust strategies that segregate patients into molecular groups useful for treatment decision and with high reliability constitute an unmet clinical need
[5].
Large-scale proteomics enabled clear separation of cell lines representing GCB and ABC subtypes
[6][7]. Follow-up clinical studies on FFPE tumor tissues
[8][9][10] have provided promising results. Nevertheless, the advantage of proteome profiling over gene expression profiling based on FFPE tumor tissue is still unclear. The potential of EV proteomes to discriminate between GCB and ABC remains to be investigated. The possibility to diagnose and screen DLBCL patients for treatment based on liquid biopsies by targeting the proteome of extracellular vesicles (EVs) is unexplored and provides clear advantages. Minimally invasive liquid biopsy-based diagnosis and patient screen opens new avenues for fast and longitudinal follow up for minimal residual disease assessment
[11][12].
EVs have attracted much recent interest because of their potential functions, use as disease biomarkers, and possible therapeutic exploitation
[12][13]. EVs are extracellular vesicles of endosomal and plasma membrane origin released in vivo into the extracellular environment by cells as distinct as B lymphocytes and dendritic cells as well as from several additional cell types of hematopoietic origin
[14]. EVs have been shown to play fundamental roles in intercellular communication by carrying and selectively deliver bioactive molecules (e.g., lipids, proteins, and nucleic acids). EVs are tailor-made specialized mini-maps of their cell of origin and of both physiological and pathological relevance
[11]. This feature leads to the potential to use EVs as blood-based biomarkers of diagnosis and prognosis for hematopoietic cancers.
2. Analysis of DLBCL Cell Lines Derived EVs
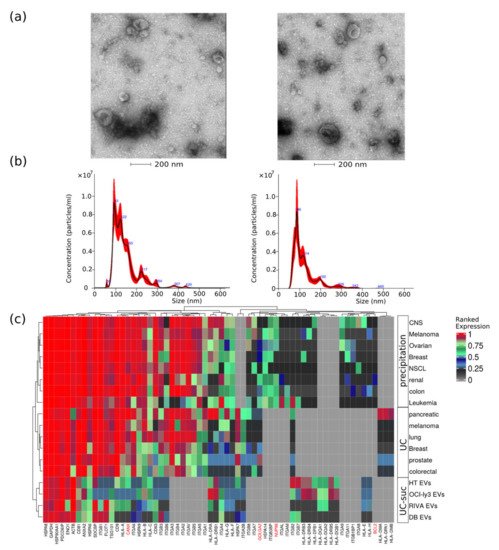
DLBCL EVs were characterized for size distribution, concentration, protein content, presence of contaminants, and morphology. By transmission electron-microscopy negative staining, we have observed vesicles with an artificial cup-shaped-like structure caused by fixation and dehydration during mounting preparation
[14][15], suggesting that EVs preparations were considerably enriched in exosomes and microvesicles with an average size between 50 and 200 nm (
Figure 1a). Size and concentration of EVs were also determined using nanoparticle tracking analysis and ranged from 80 to 240 nm (
Figure 1b) and 0.4–1.0 × 10
9 particles per microgram of protein and 1–3 × 10
11 particles per milliliter (
Table 1).
Figure 1. EVs quality control assessment evaluated by different methods. (a) Negative staining transmission electron microscopy (TEM); (b) nanoparticle tracking analysis of representative EV samples; and (c) analysis of EVs protein expression values obtained from different cancer cell types using three isolation methods, namely, precipitation, ultracentrifugation (UC), or UC combined with sucrose cushion (UC-suc). Cell organelle markers are indicated in red.
Table 1. EVs sample characteristics recorded by nanoparticle tracking analysis.
| CELL LINE. |
Mean Size (nm) |
Mode (nm) |
Particles/mL |
| DB |
141.0 ± 2.0 |
105.7 ± 6.0 |
3.07 × 1011 ± 1.89 × 1010 |
| HT |
146.0 ± 7.1 |
98.7 ± 11.3 |
1.05 × 1011 ± 8.6 × 109 |
| RIVA (RI-1) |
120.1 ± 6.6 |
81.5 ± 4.2 |
2.22 × 1011 ± 5.1 × 109 |
| OCI-ly3 |
126.9 ± 4.8 |
109.5 ± 9.5 |
1.46 × 1011 ± 1.46 × 1010 |
This corresponds to particle-to-protein ratio values concordant with the ratios reported by Webber and Clayton
[16]. To further access quality of DLBCL EVs isolations, we compared our MS data with previously published EVs MS studies using our previous developed quality assessment tool (
Figure 1c)
[12]. We observed abundant expression of known EV markers comparable to previous large-scale MS-based EV characterization studies
[17][18]. Additionally, lower-ranked protein expression levels of markers indicating contamination from other subcellular fractions such as the endoplasmic reticulum (CANX), golgi apparatus (GOLGA2), mitochondria (BCL2), and nucleus (NUP98) were in the same range as reported by others
[17][18].
3. Overview of Identified Proteins
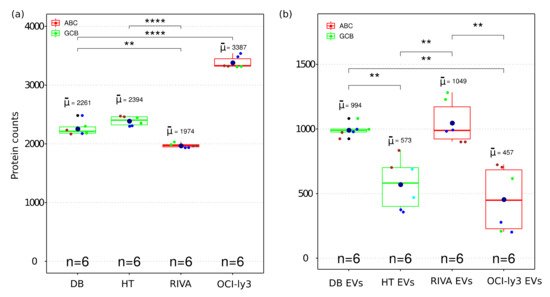
MS-based proteomics analysis of DLBCL whole-cell proteome and secreted extracellular vesicles identified in total 17,833 protein isoforms at a 1% FDR threshold. Collapsing the protein isoforms into their encoding genes resulted in 5949 proteins identified in all independent biological and technical replicates. Given the high level of replication (two technical replicas and three independent cell cultures), the statistical differences were accessed for the identified proteins across all cell lines. The total number of proteins within each cell line was highly reproducible considering both biological and technical replicates (
Figure 2a). A significant difference was observed among identified proteins between GCB and ABC subtypes. OCI-ly3 displayed a significantly higher number of identified proteins compared to the other cell lines (significant difference only indicated for OCI-ly3 in comparison with DB, in
Figure 2a). For all other comparisons, the significant differences in the number of identified proteins are indicated in
Figure 2a. RIVA displayed lower total number of identified proteins than the GCB cell lines. Proteins identified uniquely in OCI-ly3 cell line (902 proteins) were enriched in the gene ontology cellular components: nucleolus (
p value = 1.6 × 10
−12), nucleoplasm (
p value = 3.4 × 10
−7), and mitochondrion (
p value = 5.7 × 10
−7). This result pinpoints high heterogeneity in DLBCL and is concordant with previously described heterogeneity based on genomic data
[19]. It further highlights the potential of large-scale proteomics in further subclassifying DLBCL.
Figure 2. Boxplot of the number of identified proteins in whole cell (a) and EV (b) proteome from four DLBCL cell lines representing GCB and ABC sub types. The boxes indicate the median, second and third quartile. Mean values are represented by the large dark blue circles, while data points representing individual LC-MSMS runsare represented by smaller circles. All biological replica are depicted by a different small circle color and technical replica by identical small circle color. (**) correspond to p-value <0.01. (****) correspond to p-value < 0.0001.
All DLBCL cell lines analyzed the number of identified proteins from EVs and were smaller than the corresponding identifications obtained from the cellular proteome (Figure 2b). Since EVs constitute subfractions of cells, it is expected that the EVs proteome content is a subpart of the cellular proteome. The number of identifications in EVs displayed larger variance than the cellular proteomes. This can also be a result of the larger number of steps required for isolation of EVs. Nevertheless, even for EVs the numbers of identified proteins were sufficiently reproducible to allow for the detection of significant differences between EVs obtained from different cell lines.
4. Functional Enrichment Analysis of Identified Proteins
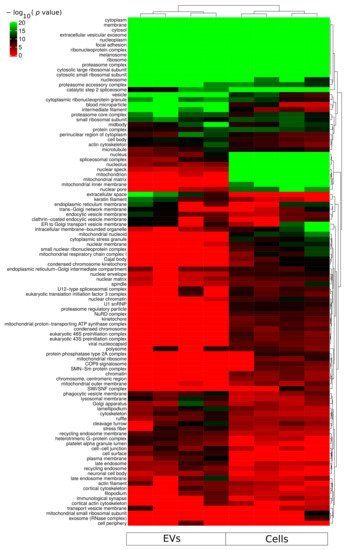
Gene ontology cellular component based functional enrichment analysis of all identified proteins revealed clear clustering patterns of cellular compartment associated proteins specific for either EVs fractions or whole-cellular proteome (
Figure 3). Cellular compartments such as extracellular vesicular exosomes, cytosolic, membrane, nucleoplasm, and nucleosome were highly enriched in both EVs and whole-cell proteomes. Proteins belonging to mitochondria-related compartments such as mitochondrion, mitochondrial matrix, and mitochondrial inner membrane were exclusively enriched in the whole-cell proteome. On the other hand, EV fractions were most enriched for proteins belonging to membrane-vesicle-related compartments such as phagocytic vesicle membrane, lysosomal membrane, recycling endosome membrane, and late endosome. In addition, while proteins of the proteasome complex were enriched in EVs, proteins of the regulatory particle 19S were less enriched in the vesicle fractions compared to the whole-cell proteome (
Figure 3). An important aspect relevant for endosomal EV sorting of protein components are ubiquitin and ubiquitin-like modifiers; and therefore, the proteasome complex is of particular relevance. The identification of proteasome complex proteins in EVs has previously been described as extracellular
[20]. However, further studies are required to assess luminal or outer membrane proteasome localization. Besides the nucleoplasm and nucleosome, all other nuclear subcellular components were mainly identified in the whole-cell proteome.
Figure 3. Functional enrichment for cellular component using all identified proteins from each sample group (cells and sEVs). A hypergeometric p-value threshold of 10−6 of at least one sample group was applied for construction of the heat map.
 +1 point
+1 point