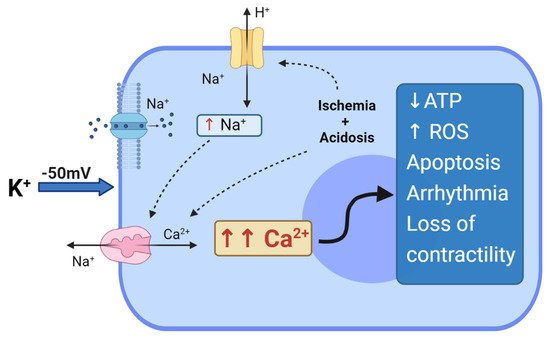
The concept of “polarized arrest” is attractive because maintaining the membrane potential close to the resting potential value (of about −80 mV) reduces the untoward effects of Ca
2+ loading. At resting membrane potentials, Na
+ and Ca
2+-channels are both closed and the transmembrane fluxes are minimized; thus intracellular, Ca
2+ overload is avoided, mitochondrial function is preserved and ATP balance maintained
[28][29]. Based on these theoretical considerations, polarized arrest should confer improved myocardial preservation and recovery, avoid oxidative stress, cell death and endothelial activation during the reperfusion period. A polarised tissue is also more resistant to ischemia and inflammations reported by several in vitro and animal studies
[30][31][32]. Indeed, when “inflammatory-resistance” is considered, polarizing solutions have been reported to have a greater effect on reducing porcine neutrophil priming through a greater suppression of superoxide anion generation
[30]. Granfeldt and colleagues also confirmed that ALM infusion in pig models induces a reversible hypotensive and hypometabolic state, attenuates tumor necrosis factor-α levels and improves cardiac and pulmonary function
[31]. Liu et al. also reported how adenosine adjunct to blood caridoplegia results in a lower troponine leakage (ischemia) as well as IL-6 secretion (inflammation)
[32].
All of these findings refer to animal or in vitro studies, but it is reasonable to think that the membrane polarization has similar effects in human myocytes, thus providing superior myocardial protection.
2.1. From the Isolated Heart to Animal Studies:
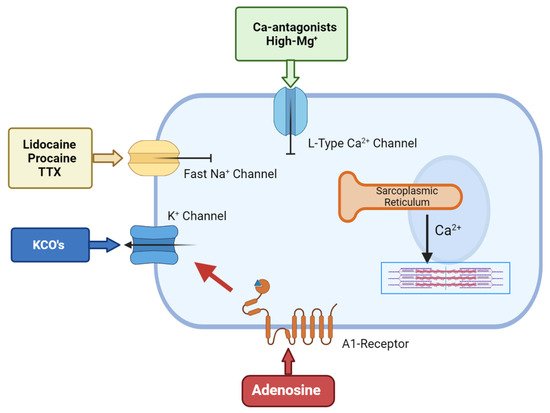
Polarized arrest can be achieved using a number of drugs. The most common are the Na+-channel blockers such as lidocaine, procaine and tetrodotoxin (TTX) and these have been used as cardioplegic adjuncts for many years. All three block the voltage-gated Na+ fast channels that are responsible for the action potential upstroke, thereby maintaining the membrane potential at or around its resting value
[33][34][35] (
Figure 2). Lidocaine or procaine is often used as an additive in Bretschneider and STH solutions
[36][37]. Lidocaine (and procaine) alone have not been translated as primary cardioplegic agents
[38] because of their significant adverse effects (i.e., torsade de pointes and convulsions in from 1% to 8.6% during local and regional anhestesia)
[34]. Similarly, although TTX has been shown to improve the post-ischemic recovery in rat hearts compared to hearts that were arrested using high K
+ solutions, the toxicity of the drug prevents it from use in cardiac surgery
[39][40][41][42][43].
Figure 2. The cellular targets for hyperpolarized/polarized arrest.Adenosine-related cardioplegic effect arises from the inhibition of the sino-atrial and atrioventricular nodes, as well as from the activation of A1 receptors located on atria and ventricular cells, which open sarcolemma KATP channels to reduce action potential duration. KCO’s allows an increased potassium flux through the sarcolemma that will shift membrane potential towards the K+ equilibrium potential, which is normally around −90 mV in myocytes. Local anesthetic agents block the voltage-gated Na+ fast channels that are responsible for the action potential upstroke, thereby maintaining the membrane potential at/around its resting value. Ca-antagonist agents inhibit the L-type calcium channels, thus inhibiting the smooth muscle cell’s contractility.
Another family of polarising compounds discovered in the early 1990s are the ATP-sensitive K
+ (K-
ATP) channel openers
[44] (
Figure 2). Sarcolemma K
ATP channels are present in most tissues including heart and brain
[34] and, upon activation, protect the cell against ischemia-reperfusion injury by lowering intracellular Ca
2+ and preserving ATP
[45][46]. At higher concentrations, K
ATP openers shorten the action potential duration and arrest the heart at hyperpolarized potentials
[47][48]. A number of K
ATP-channel openers have been synthesized, including nicorandil, aprikalim, pinacidil, and cromakalim
[28]. Cohen and colleagues compared the cardioprotective effect of aprikalim to a hyperkalemic solution in isolated crystalloid-perfused rabbit hearts subjected to 20 min normothermic global ischemia. Hearts arrested with aprikalim recovered significantly better compared to hearts arrested by high K
+ solution
[49]. Unfortunately, like many new drugs, the application of K
ATP openers failed to translate from the high incidence of reperfusion arrhythmias (about 60% in animal models)
[44][45][46][47][48][49].
A third alternative to high K
+ involves non-depolarizing esmolol-HCl cardioplegia
[28][50][51][52]. The idea was pioneered by Chambers and colleagues, who showed that 1 mmol/L esmolol induced a diastolic cardiac arrest in isolated rat hearts, and in the presence of oxygenated perfusate, recovered faster compared to cross-clamp fibrillation or STH hyperkalemic cardioplegic solution
[53][54]. Esmolol is an ultra-short-acting cardioselective β1-blocker that inhibits the L-type calcium channels and fast Na
+ channels, and produces a pronounced negative inotropy, slows conduction and, at high concentrations, induces polarized arrest
[55][56][57][58]. However, at high concentrations, Pirk and colleagues reported prolonged infusion periods (beyond 20 min) may compromise its reversibility
[56]. More recently, Chambers and colleagues reported lower concentrations of esmolol (0.6 mM) combined with adenosine (0.25 mM) alleviates this problem, with significantly improved protection compared to STH solution
[57][58]. The combination of esmolol and adenosine may provide a clinically relevant polarizing cardioplegia, although further studies are required.
Adjunctive adenosine has a long history in cardioplegia, beginning in the late 1980s. Belardinelli paved the way by demonstrating that adenosine induced hyperpolarized arrest in isolated sino-atrial (SA) node pacemaker cells
[59]. This ‘cardioplegic’ effect was confirmed by Schubert et al., who compared 10 mM adenosine alone to hyperkalemic solutions in isolated rat hearts
[60]. De Jong and colleagues further confirmed that high levels of adenosine induced rapid arrest; however, they also showed it delayed post-ischemic recovery, which seriously hampered its translation to humans
[61]. Adenosine’s cardioplegic effect arises from inhibition of the SA and atrioventricular nodes and via activation of A1 receptors located on atria and ventricular cells, which open sarcolemma K
ATP channels to reduce action potential duration
[2] (
Figure 2).
A question that has not been adequately addressed in the literature is why adenosine or mechanistic K
ATP openers have failed to translate into an effective polarising cardioplegia. In 1998, Dobson proposed that the reason for this was related to the inability of each drug to alter the upstroke of the action potential
[2]. Reducing the action potential duration will arrest the heart, but, alone, it is insufficient for a coordinated return to sinus rhythm. Dobson instead proposed that combining adenosine with lidocaine may be more effective for the following: (1) shortening the action potential, and (2) inhibiting Na
+ fast channels, which flatlines the action potential and arrests the heart at polarised potentials
[2]. Magnesium was added to confer additional stability for arrest and reanimation. The Adenosine–Lidocaine–Magnesium (ALM) solution concept was borrowed from natural hibernators and developed at James Cook University, Australia
[62]. The idea received proof-of-concept in isolated working rat heart preparations, followed by translation to a canine cardiopulmonary bypass model
[2]. Dobson and colleagues also demonstrated the safety and superiority of ALM as a cardioplegia compared to traditional hyperkaliaemic solutions, as well as a preserving a solution even after 8 hours of cold static storage
[63]. Further studies confirmed that ALM not only protects the myocardium, but also significantly reduces coronary vasculature resistance, inflammatory response, lung oedema, and prevents coagulopathy
[64][65][66][67][68][69].
2.2. Humans Trials: Are We Getting Closer to Change?
It is remarkable that, despite 60 years of experimenting with cardioplegic solutions, there are only a handful of clinical experiences on fully-polarizing cardioplegia. Most trials have been with fully depolarising potassium solutions, with polarizing agents as additives rather than as the primary arresting agents. For example, Liu and colleagues compared standard cold blood K
+ cardioplegia in heart valve replacement to adenosine pre-treatment plus the cardioplegia, and reported a reduced Troponin-I, IL-6 and IL-8 release and reduced myocardial injury after adenosine pre-treatment
[32]. Mentzer and colleagues found a lower incidence of inotropic support in patients receiving adenosine as an additive to hyperkalemic blood cardioplegia compared with controls (the 24-h average doses for dopamine and nitroglycerine in the placebo group were 28-fold and 2.6-fold greater than their respective high-dose adenosine treatment cohorts), suggesting a better myocardial recovery in patients undergoing CABG
[70]. More recently, Abdelwahab and colleagues showed that the use of adenosine immediately after aortic cross-clamping and prior to the infusion of cold hyperkalaemic cardioplegia significantly decreased postoperative Troponin-I leakage compared to standard hyperkaliemic cardioplegia alone
[71]. Our Institution also conducted a trial on patients undergoing urgent CABG, and we demonstrated improved myocardial protection and functional recovery after cardioplegic arrest with the addition of polarizing ALM to a standard hyperkalemic cold blood vial
[72].
Baraka and colleagues used lidocaine addition (100 mg/L) to crystalloid cardioplegic solution to prevent ventricular fibrillation after the release of the aortic cross-clamp in 50 patients undergoing CABG and in 30 patients undergoing mitral or aortic valve replacement. In the coronary artery bypass grafting group, lidocaine cardioplegia significantly reduced the incidence of reperfusion ventricular fibrillation from 100% to 42%. In the valve group, lidocaine cardioplegia also significantly reduced the incidence of reperfusion ventricular fibrillation from 93% to 42%. They reported that lidocaine cardioplegia decreased the number of direct-current countershocks required to defibrillate the heart, with no significant increase in the incidence of high-grade atrioventricular block
[73]. In another trial, Ramani and colleagues compared their modified single-dose, long-acting, lidocaine-based blood cardioplegia with short-acting STH1 blood cardioplegia in patients undergoing single valve replacement. The group concluded that the method was safe and efficacious; however, there was no significant difference in creatine-phosphokinase-MB (CK-MB), Troponin-I levels, lactate level and myocardial recovery, and the study lacked statistical power to support their conclusion
[74]. Finally, another randomized trial compared cardioplegia with and without lidocaine in patients undergoing CABG. Lidocaine-enriched cardioplegia significantly reduced the incidence of reperfusion ventricular fibrillation from 63% to 42%, while the incidence of atrioventricular block was higher in the lidocaine group
[75].
Procaine is another local anesthetic with a long history as an additive to standard cardioplegic solutions. Mustonen et al. showed that patients undergoing CABG receiving procaine in cardioplegia had a shorter mean ventricular fibrillation time (27 ± 79 s vs. 205 ± 161 s,
p < 0.001) and achieved stable rhythm in a higher proportion (67.6% vs. 13.5%,
p < 0.001). Moreover, the mean number of defibrillations was lower than in patients receiving placebo (0.3 ± 0.7 vs. 2.4 ± 1.7,
p < 0.001)
[76]. Similar results were reported by Sellevold et al., who added 1mM procaine to STH2 cardioplegia in patients undergoing CABG compared to the same solution with saline adjunct. The number of synchronized direct-current shocks for conversion of atrial fibrillation was lower in the procaine group (2% vs. 100%,
p < 0.001), as well as the post-operative myocardial enzymatic release
[77].
Potassium channel openers, such as nicorandil, added to high K
+ solutions have also been trialled in open heart surgery. Hayashi et al. compared the peri-operative results of cardioplegia with nicorandil adjunct to standard cardioplegia in patients undergoing elective CABG. The time required to achieve cardiac arrest after the initiation of cardioplegia was significantly reduced in the nicorandil group, as well as ECG-based ischemic signs during reperfusion. Furthermore, the recovery of sinus rhythm was significantly greater for nicorandil group
[78]. Similar results have been showed by Chinnan et al., who added nicorandil to hyperkalaemic solution during mitral valve and CABG surgery. He reported that nicorandil did not cause significant haemodynamic changes or malignant arrhythmias in any patient
[79]. Another study used nicorandil as pre-treatment before cardioplegia, confirming an enhanced myocardial protection
[80].
Esmolol-based cardioplegia has been examined in a randomized single-center trial by Scorsin and colleagues on 41 patients scheduled for isolated aortic valve replacement to continuous coronary infusion with either K
+ or esmolol during CPB
[81]. Coronary glucose and lactate transmyocardial gradients were similar in both groups, indicating adequate myocardial perfusion in all patients. It was further suggested that esmolol could be effective for myocardial protection in hypertrophied hearts by reducing myocardial oxygen metabolism
[81]. In another randomized single-centre trial, esmolol was used as adjunct to high K
+ cardioplegia and it enhanced postoperative cardiac performance and reduced the postoperative troponin leakage in high-risk patients undergoing elective cardiac surgery
[82][83]. In contrast, Rinne and colleagues showed that the addition of esmolol to blood cardioplegia did not increase the efficacy of cardioprotection in unstable patients during urgent coronary revascularization
[84]. Therefore, the data on esmolol are conflicting and suggest further investigations in clinical practice.
In contrast, ALM cardioplegia has shown increasing promise as a fully polarizing cardioplegia in a number of human trials. The first clinical trial was conducted by Jin and colleagues involving 134 pediatric patients with low-risk congenital heart disease. The results showed cold AL crystalloid cardioplegia in pediatric patients was safe and associated with higher postoperative systolic pressures, lower blood troponin-I levels and a shortened hospitalization stays, compared with hyperkalaemic cardioplegia
[85]. Jakobsen and colleagues
[86] also reported that adenosine instead of high K
+ in cold crystalloid cardioplegia (supplemented with procaine) was safe, provided a more rapid cardiac arrest, and afforded similar cardioprotection in sixty patients undergoing CABG
[87]. They also reported that their adenosine and procaine solution resulted in similar hemodynamic parameters as high K
+ but with a marked reduction in the incidence of postoperative atrial fibrillation by more than 50%
[88].
To our knowledge, the only randomized human trial on full-polarizing ALM cardioplegia was conducted at our Institution in Verona Medical School. In 2016, we documented, for the first time, that “full-polarizing” ALM at high doses in a normokalemic cold blood vial was superior in humans in terms of intraoperative myocardial anaerobiosis and postoperative myocardial enzymatic leakage in both elective CABG and valve surgery, when compared to the Buckberg depolarizing cold blood solution
[89]. Since then, ALM cold blood cardioplegia has been widely used at our Institution for elective adult cardiac surgery. We recently collected data from one-thousand consecutive elective adult cardiac patients (627 undergoing ALM-polarizing cardioplegia vs. 373 Buckberg cardioplegia) operated upon at our Institution over a 20-month period. These data (unpublished) confirmed significantly lower leakage of high-sensitivity troponin I (TnI
max: ALM-POL-group: 9796 ± 8888 ng/L vs. BUCK-DEPOL-group: 12,879 ± 10,362 ng/L) during hospitalization as well as a higher spontaneous recovery of sinus rhythm at aortic declamping in ALM-group (70.9% vs. 50.9%,
p < 0.001). These data confirm the safety of a full-polarizing cardioplegia and that the ALM solution is a strong contender for the first clinically proven alternative to high K
+ solutions. This may be timely given that our future patients will be older, and sicker with multiple comorbidities. The future of polarized cardiac arrest is an exciting one and may play a crucial role in daily cardiac surgical practice.
 +1 point
+1 point