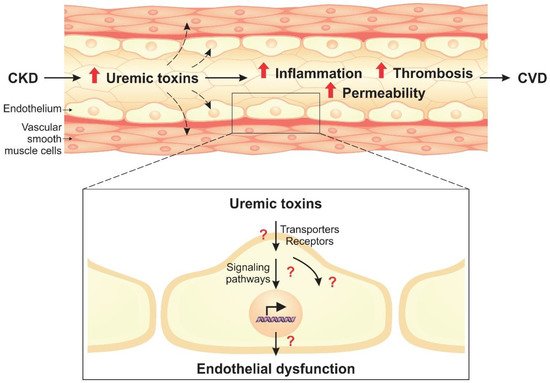
Uremic toxins lead to phenotypic endothelial changes through the activation of cellular signaling pathways. Given the complexity of these pathways, important proteins that modulate the cellular response to uremic toxins stand out, such as AhR, NF-κB, and MAPKs. These molecules, therefore, have potential to be used as therapeutic targets in CKD.
2.1. AhR Pathway
AhR is a ligand-inducible transcription factor that belongs to the basic helix–loop–helix transcription factor family. Inactivated AhR is found in the cytoplasm bound to chaperones, which dissociate when AhR is activated by binding to a ligand
[15]. Studies have identified several exogenous and endogenous ligands for AhR, such as 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD), dioxin-like planar polychlorinated biphenyls (PCBs), and uremic toxins, including IS, IAA, indoxyl glucuronide, kynurenic acid, and other tryptophan derivatives
[16][15][17][18][19][20][21]. Activated AhR can induce genomic signaling, known as the canonical pathway, as well as the non-genomic pathway.
In the genomic signaling pathway, activated AhR is translocated to the nucleus, where it forms a dimer with aryl hydrocarbon receptor nuclear translocator (ARNT)
[15]. The AhR/ARNT complex binds to the xenobiotic responsive element (XRE; 5′-GCGTG-3′) in the promoter region of several target genes, inducing their expression
[15][22]. Studies have shown that IS and IAA induce AhR translocation to the nucleus in endothelial cells
[23][16][17]. In addition, overexpression of
CYP1A1 and
CYP1B1, genes directly regulated by AhR/ARNT, was found in endothelial cells exposed to IS and IAA, an effect that was reversed with AhR inhibitors
[23][16][24][17]. Furthermore, it has been demonstrated in HUVECs that IS and IAA, through AhR, induce AhRR expression, an AhR repressor that competes for ARNT binding, which ultimately leads to a negative regulatory loop for AhR
[25][24][22].
In the non-genomic pathway, activated AhR interacts with various other signaling molecules, such as NF-κB and Src, independently of ARNT
[16]. Ito et al.
[23] demonstrated that IS increased the expression of E-selectin in an AhR-dependent manner in HUVECs. Despite that, the authors verified that AhR did not directly bind to the E-selectin gene promoter
[23]. However, it was found that E-selectin overexpression was associated with the activity of the transcription factor activator protein-1 (AP-1), which is induced by AhR through the non-genomic pathway
[23]. Similarly, Addi et al.
[16] demonstrated that HUVECs exposed to IAA had an increase in TF gene expression, but AhR was not linked to the gene promoter despite the effect being reversed with AhR knockout and inhibition. The authors then found that NF-κB was essential for increasing TF expression, but its activity was decreased by the AhR inhibitor, suggesting a regulation between them
[16].
Studies have shown that the activation of AhR by uremic toxins is involved in vascular inflammation, permeability, and the development of CVD
[23][26][17]. In HUVECs, IS induced MCP-1 and E-selectin expression, proinflammatory molecules involved in leukocyte recruitment and adhesion to the endothelium, in an AhR-dependent manner
[23][17]. In vivo, Ito et al.
[23] also found that IS increased interaction between leukocytes and the endothelium of the femoral artery in wild-type mice, an effect that was not seen in endothelial cell-specific AhR knockout mice treated with IS. Furthermore, the activation of AhR by IS also enhanced Src phosphorylation and, consequently, VE-cadherin phosphorylation, inducing an increase in endothelial permeability that was reversed with AhR inhibitors
[26]. Koizumi et al.
[27] demonstrated that IS-induced senescence of HUVECs is AhR-dependent and may contribute to CVD. In HUVECs, IAA and IS increased the expression of TF through AhR, which is associated with the pathogenesis of atherosclerosis and thrombosis
[25][16]. Therefore, these studies suggest that AhR activation induced by uremic toxins has an important role in endothelial dysfunction and vascular injury. Interestingly, IS through the AhR pathway led to upregulation of OAT1 in renal proximal tubule cells as well as P-glycoprotein, an efflux pump that is part of the ABC transporter superfamily, in human hepatoma cells
[28][29]. These data suggest that the AhR pathway may be involved in the regulation of the expression of cellular transporters.
AhR activating potential (AhR-AP) corresponds to the combination of all AhR agonists present in uremic serum, such as IS, IAA, and indoxyl glucuronide. Dou et al.
[30] demonstrated that uremic serum from stage 3 to 5 and stage 5D CKD patients had higher AhR-AP than serum from healthy controls by an AhR-responsive bioassay. In addition, the authors reported that AHR-AP is associated with cardiovascular events in CKD patients
[30]. In vivo, Dou et al.
[30] detected higher serum levels of AhR agonists as well as overexpression of
Cyp1a1, a gene regulated by AhR, in the aorta and heart of nephrectomized mice compared to AhR
-/- nephrectomized or wild-type mice. In a cohort of patients with ESRD, Shivanna et al.
[31] noted greater AhR activity in uremic serum compared to that of healthy controls. Kolachalama et al.
[21] also found increased AhR activity and TF levels in serum from CKD patients who had thrombotic events compared to their counterparts without thrombosis. Taken together, these data indicate a relationship between uremic toxins, AhR activation, and the development of CVD
[21][30][32].
2.2. NF-κB Pathway
NF-κB is a family of transcription factors that play a crucial role in endothelial inflammation. There are five forms of NF-κB proteins: p50 (NF-κB1), p52 (NF-κB2), p65 (RelA), RelB, and c-Rel. The p52 and p50 proteins are derived from the p100 and p105 forms, respectively. NF-κB family members form dimers, of which the p50/p65 dimer is the most common
[33][34]. In the inactivated state, NF-κB dimers are found in the cytoplasm bound to inhibitory proteins, such as IκBα and IκBβ. In the presence of a stimulus, NF-κB dissociates from the inhibitory proteins, which are degraded. In the canonical pathway, the activation of IκB kinase (IKK) mediates the phosphorylation of IκBα, releasing NF-κB (mainly the p50, p65, and c-Rel forms) which is then translocated to the nucleus
[33][34]. On the other hand, in the non-canonical pathway, also known as the alternative pathway, the activation of NF-κB-inducing kinase (NIK) leads to p100 phosphorylation and processing to the p52 form, which is translocated to the nucleus along with RelB
[33][34].
Studies have shown that the NF-κB pathway participates in gene regulation in endothelial cells in the uremic conditions
[35][24][36]. HUVECs exposed to uremic serum had greater IκB degradation and, consequently, higher levels of p50/p65 in the nucleus compared to those exposed to healthy serum
[37]. In HUVECs, Tumur et al.
[36] demonstrated that IS induced p65 phosphorylation as well as MCP-1 and ICAM-1 overexpression, which was reversed with NF-κB inhibitors. Furthermore, Masai et al.
[35] reported an increase in the translocation of p65 to the nucleus and MCP-1 upregulation in HUVECs exposed to IS. Interestingly, the authors verified that this effect was suppressed by ERK1/2 and p38 MAPK inhibitors, indicating the participation of these proteins in the activation of the IS-induced NF-κB pathway
[35]. In addition, studies have demonstrated an increase in the translocation of p50 and p65 to the nucleus in HUVECs exposed to IAA, which was decreased with AhR and p38 MAPK inhibitors
[16][24]. Inhibition of NF-κB also reversed IAA-induced COX-2 and TF overexpression in HUVECs
[16][24]. Based on these data, it is suggested that the NF-κB pathway is important for the upregulation of proinflammatory proteins in uremic conditions.
2.3. MAPK Pathway
MAPK are a family of serine/threonine kinases that are activated by phosphorylation
[38]. There are three main groups of MAPK: Extracellular signal-regulated kinase (ERK1 and ERK2), C-Jun N-terminal kinase (JNK1, JNK2, and JNK3), and p38 MAPKs (α, β, γ, and δ)
[39]. The MAPK signaling pathway is activated in the presence of a stimulus and, consequently, mediates the cellular response through activation of other proteins, such as transcription factors, by phosphorylation
[40][41]. Therefore, MAPK has a significant role in signal transduction
[39].
In uremic conditions, studies have shown that the MAPK pathway is activated in endothelial cells. Uremic serum enhanced ERK1/2 phosphorylation in HUVECs compared to normal serum
[37]. IAA and IS also induced phosphorylation of p38 MAPK and ERK1/2 in HUVECs in the first 30 min of exposure
[35][24]. In addition, MAPK pathway inhibition can reverse the effects of uremic toxins
[35][16][24]. Inhibition of p38 MAPK reduced IAA-induced TF and COX-2 overexpression in HUVECs, the expression of which are possibly modulated by the AhR/p38MAPK/NF-κB pathway
[16][24]. In another study, it was shown that inhibition of ERK1/2 and p38 MAPK suppressed IS-induced p65 phosphorylation (NF-κB) as well as MCP-1 overexpression in HUVECs
[35].
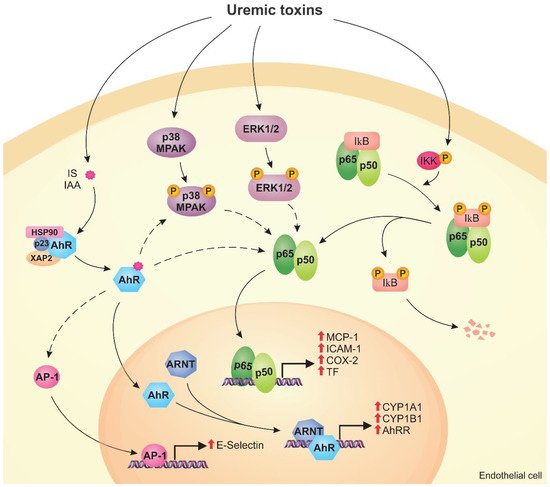
Figure 2 shows the mechanisms modulated by the AhR, NF-κB, and MAPK signaling pathways in endothelial cells exposed to uremic toxins.
Figure 2. Uremic toxins activate the aryl hydrocarbon receptor (AhR), nuclear factor kappa B (NF-κB), and mitogen-activated protein kinase (MAPK) pathways in endothelial cells. Toxins such as indoxyl sulfate (IS) and indole-3 acetic acid (IAA) activate AhR, which is translocated to the nucleus, forms a dimer with aryl hydrocarbon receptor nuclear translocator (ARNT), and induces the expression of CYP1A1, CYP1B1, and AhRR (genomic pathway). Activated AhR can stimulate other pathways (non-genomic pathway), including the activation of AP-1 that induces E-selectin expression. Uremic toxins also cause the activation of the MAPK pathway, such as p38MAPK and ERK1/2, by phosphorylation. MAPK and AhR can induce the NF-κB pathway. In uremic conditions, IκB is degraded and p50/p65 are translocated to the nucleus, inducing monocyte chemoattractant protein-1 (MCP-1), intercellular adhesion molecule-1 (ICAM-1), cyclooxygenase-2 (COX-2), and tissue factor (TF) expression. The activation of these pathways by uremic toxins can lead to endothelial dysfunction.
 +1 point
+1 point