The ryanodine receptor (RyR) is a Ca2+ release channel in the sarcoplasmic reticulum of skeletal and cardiac muscles and plays a key role in excitation–contraction coupling. The activity of the RyR is regulated by the changes in the level of many intracellular factors, such as divalent cations (Ca2+ and Mg2+), nucleotides, associated proteins, and reactive oxygen species. Since these intracellular factors change depending on the condition of the muscle, e.g., exercise, fatigue, or disease states, the RyR channel activity will be altered accordingly.
1. Introduction
Skeletal and cardiac muscles are essential organs for the maintenance of life, which embody most of the daily activities, such as walking, eating, and creating. Skeletal muscles are required to perform flexible and complex movements, generate and maintain force, and respond rapidly to different conditions, while the heart muscle sustains and regulates heartbeat according to physical activity. In addition, during human physical activities, muscles are subjected to high-loads and fatigue and various external stimuli, such as injury, stress, and excitement. These external stimuli cause intricate changes in the intracellular environment of muscle cells. The muscle can sense these intrinsic changes and immediately respond to them.
The contractile activity of skeletal and cardiac muscles is induced by a series of events, including action potentials, Ca
2+ release from the sarcoplasmic reticulum (SR), and contractile force generation. The ryanodine receptor (RyR) is a Ca
2+ release channel in the SR. In skeletal muscle, the type 1 RyR (RyR1) interacts with the dihydropyridine receptor (DHPR or Cav1.1), an L-type Ca
2+ channel in the transverse tubule (T-tubule). As a result, the RyR1 channel opening is triggered by a conformational change in the DHPR after depolarization of the T-tubule (referred to as depolarization-induced Ca
2+ release, DICR)
[1][2][3]. In cardiac muscle, the type 2 RyR (RyR2) is opened by the binding of Ca
2+ that is introduced from the extracellular space through the DHPR (Cav1.2) (referred to as Ca
2+-induced Ca
2+ release, CICR). However, in cardiac muscle, a direct interaction between the RyR2 and DHPR is lacking. The opening of RyR channels releases massive Ca
2+ from the SR. Thus, the intracellular Ca
2+ concentration rapidly increases from 50 nmol/L during the resting state to 10–20 μmol/L as a maximum peak in twitch contraction
[4][5][6]. A structural change in tropomyosin on the thin muscle filament is caused by Ca
2+ binding to troponin C, and this change facilitates the interaction between myosin and actin, myosin ATPase activity and the sliding of the two filaments, resulting in muscle contraction
[7][8]. Thus, the RyR is one of the most important elements in excitation–contraction coupling and a central component for the myocyte function.
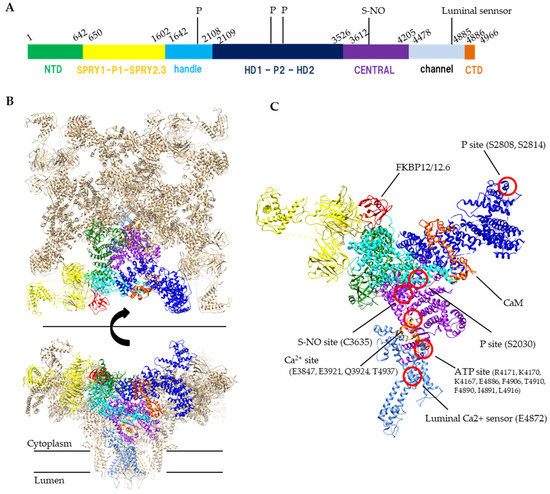
The RyR is a huge (>2 MDa) protein complex, which is composed of four 550 kDa subunits. The RyR is a six-transmembrane P-type channel at the C-terminus with a large N-terminal cytoplasmic region which covers 70% of the entire molecule
[9]. Recent cryo-EM studies of RyRs revealed a complex structure with over 15 domains
[10][11][12]. The transmembrane S1, S3 and S5 helices penetrate the SR membrane from the cytosolic side to the luminal side, whereas the S2, S4 and S6 helices penetrate the membrane in the opposite direction. The S6 helix forms a pore for Ca
2+ ions. The S2-S3 domain, which is the region between S2 and S3 helices on the cytosolic side, is thought to be involved in regulation of channel opening. The S4–S5 linker is a short helix that runs parallel to the SR membrane between the S4 and S5 helices and participates as a stopper against the movement of S6 helix. The C-terminal domain (CTD) forms a part of Ca
2+ and nucleotide binding sites at the C-terminal side of the S6 helix. The C-terminal domain is involved in channel gating and comprises the channel core. The large N-terminal cytoplasmic region which is unique to the RyR family contains multiple domains, including the N-terminal domain (NTD), SPRY1, P1, SPRY2, SPRY3, Handle, HD1, P2, HD2, and CENTRAL domains (
Figure 1). Since many disease-linked point mutations have been identified in RyR cytoplasmic domains, this region is thought to be involved in the regulation of channel activity
[13].
Figure 1. Structure of RyR. (
A) A schematic illustration of domain organization of mouse RyR2. (
B) Structure of the tetrameric RyR2 in complex with FKBP12.6 and calmodulin, looking from the cytoplasmic side (top) or parallel to the membrane (bottom). PDB code: 6JI8. (
C) Structural details of domain organization and modification sites in one protomer. S-NO site and p site indicates s-nitrosylation site and phosphorylation sites, respectively. Domain colors correspond to a schematic illustration of A, NTD (residues 1–642) colored forest green, SPRY1-P1-SPRY2-SPRY3 (residues 650–1602) colored yellow, handle (residues 1642–2108) colored cyan, HD1-P2-HD2 (residues 2109–3526) colored blue, CENTRAL (residues 3612–4205) colored purple, channel region (residues 4478–4885) colored light blue, CTD (residues 4886–4966) colored orange. FKBP12.6 and calmodulin (CaM) are colored red and orange red, respectively.
2. Factors Affecting RyR Function
2.1. Ca2+
Ca
2+ is a primary ligand for RyRs that induces channel opening. The channel activity of all RyR subtypes increases at micromolar concentrations of Ca
2+. However, Ca
2+ does not only open the channel but also leads to channel closing when higher concentrations are reached (sub-millimolar or more)
[14][15]. The biphasic Ca
2+ dependence is explained by the presence of two distinct Ca
2+-binding sites: a high-affinity site involved in opening the RyR channel and a low-affinity site for inactivation
[16][17]. The Ca
2+ concentration for the inactivation differs between RyR1 and RyR2; the activity of RyR1 is inhibited by sub-millimolar Ca
2+; however, millimolar or more Ca
2+ level is required for the inhibition of RyR2. The high-affinity Ca
2+-binding site for channel activation is located in the interface between the Central domain and CTD
[10] (
Figure 1).
2.2. Mg2+ and Adenine Nucleotides
Mg
2+ has an inhibitory effect on the RyR channels
[17][18][19]. Mg
2+ acts as a competitive inhibitor for the activating high-affinity Ca
2+-binding site and an agonist for the inactivating, low-affinity Ca
2+ site
[20][21]. The EC
50 of Mg
2+ for the inactivating Ca
2+-binding site is similar to that of Ca
2+, whereas the EC
50 is lower than that of Ca
2+ for the activating Ca
2+ site
[22][23]. Although Mg
2+ is predicted to bind to these Ca
2+-binding sites, there is no structural evidence for Mg
2+ binding thus far.
Adenine nucleotides (ATP, ADP and AMP) increase the RyR activity
[18][19]. The rank order of activation is ATP>ADP>AMP. The EC
50 of ADP to RyR2 is one order of magnitude higher than that of ATP
[24]. The concentration of ATP, ADP and AMP at resting skeletal muscle is 8.5 mmol/L, 0.008 mmol/L and 0.007 μmol/L, whereas those after exercise is 8.5 mmol/L, 0.15 mmol/L and 2.7 μmol/L, respectively
[25]. RyR is predicted to have two ATP-binding sites in one protomer
[26], but structural studies have revealed that only one ATP is identified at the interface between Central, S6 and CTD which is near the Ca
2+ binding site
[10] (
Figure 1). Because other adenine nucleotides can also bind to the site, displacement of ATP by ADP might occur when ADP concentration is increased. In consequence, an increase in ADP concentration causes a reduction in Ca
2+ release.
The variation in the concentration of the factors described above is relevant to RyR activity in high-load exercise. We will discuss these changes further in the Exercise and fatigue section of this review.
2.3. SR Luminal Ca2+ and Calsequestrin
Ca
2+ release through RyR2 occurs spontaneously when a high luminal Ca
2+ concentration is present
[27][28][29]. Some studies have discussed the mechanism of sensing the luminal Ca
2+ concentrations in relation to ATP. Single-channel recording showed that the affinity of the RyR for ATP decreased when the concentration of luminal Ca
2+ was less than 1 mM, and the effect of ATP on RyR activation increased when the concentration of luminal Ca
2+ was 8–35 mmol/L
[30]. Single-channel recording experiments showed that the activity of RyRs increased when the concentration of luminal Ca
2+ increased above 1 mmol/L in the SR, whereas trypsin treatment of the luminal side of the SR membrane decreased RyR activity after Ca
2+ concentrations increased
[31]. Therefore, it was proposed that RyRs have at least two types of luminal Ca
2+ sensor sites in the SR lumen: one for activation and one for inhibition. A recent study of RyR2 showed that the E4872A mutation abolished luminal Ca
2+ activation, suggesting that this site may be part of the luminal Ca
2+ sensor
[32]. It was predicted by simulation that Ca-binding with the luminal region of RyR was also predicted by simulation study
[33]. In imaging studies of flexer digitorum brevis
[34] and tibialis anterior
[35] muscles, the concentration of luminal Ca
2+ was ~0.4 mmol/L at rest and fell to ~0.08 mmol/L after tetanic stimulation at 50 Hz.
Calsequestrin is a low affinity, high capacity Ca
2+-binding protein in the SR and is thought to act as a Ca
2+ buffer
[36][37][38]. In addition, calsequestrin inhibits RyR channel activity
[39][40]. It has been reported that the inhibitory effect of calsequestrin is induced when the luminal Ca
2+ concentration drops below 0.1 mmol/L
[41][42]. Moreover, calsequestrin binds to junctin and triadin, which are SR membrane proteins that promote Ca
2+ release though the RyR, and depresses the effect of these proteins
[42][43]. Conversely, calsequestrin dissociates from triadin with the increasing the luminal Ca
2+ concentration, and inhibitory effect of calsequestrin on RyR is reduced
[39][44]. The variation in the concentration of the factors described above is relevant to RyR activity in high-load exercises. We will discuss them in the
Exercise and Fatigue section of this review.
2.4. Dihydropyridine Receptor (DHPR)
In skeletal muscle, RyR1 is under the control of the DHPR. Therefore, abnormalities in the DHPR are directly related to RyR1 function
[45][46][47]. Amino acid mutations in the DHPR cause diseases such as malignant hyperthermia (MH) and myopathy
[48]. Malignant hyperthermia is a life-threatening disorder characterized by skeletal muscle rigidity and elevated body temperature in response to volatile anesthetics
[49]. Although the primary genetic causes of MH are mutations in the RYR1 gene
[50], some mutations have also been found in the genes for α
1S and β
1a subunits of DHPR
[48].
3. RyR Regulation under Different Muscle Conditions
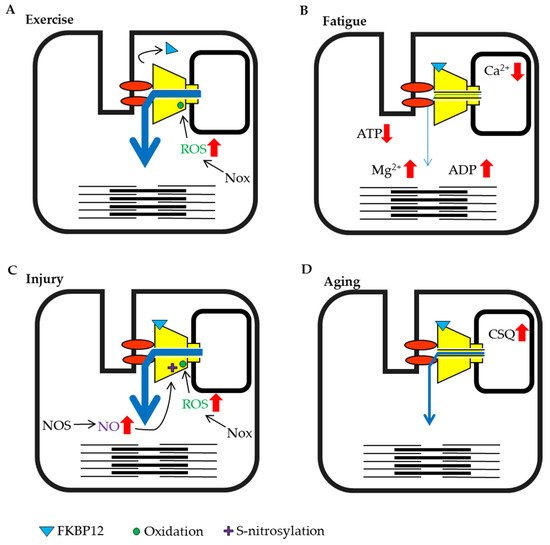
3.1. Exercise and Fatigue
Exercise and high load-bearing work cause various changes in skeletal muscle. In particular, during eccentric contraction, the expression of the NADPH oxidase (Nox)4, which produces superoxide from oxygen is increased and substantial ROS are generated
[51][52]. Nox inhibitors abolish the tetanic stimulation-induced increase in Ca
2+ release in skeletal muscle myotube
[53]. This result might indicate that exercise induced-ROS generation is involved in the acute activation of RyR1 and enhancement of muscle contraction, whereas RyR1 function is impaired if ROS continued to oxidize RyR1
[54]. It has also been reported that the binding of FKBP12 to RyRs in muscle is reduced by eccentric contraction
[55], and as noted above, dissociation reduces FKBP12-mediated suppression of RyR activity (
Figure 2A). In most cases, exercise increases RyR activity, which in turn increases muscle tension.
Figure 2. Schematic representation of skeletal muscle ryanodine receptor (RyR1) modulation by intracellular factors under different conditions. (A) Exercise training by eccentric contraction. (B) High load bearing work and fatigue. (C) Injury condition caused by damage, heat stress or disuse. (D) Aging.
When high-frequency stimulation is sustained, muscle tension gradually declines and the muscle falls into a state of fatigue. Under these conditions, a large amount of ATP is consumed by myosin and sarcoendoplasmic reticulum calcium ATPase (SERCA) in the muscle. Studies measuring ATP concentrations in creatine-kinase inhibited muscle fibers have shown that the ATP concentration in resting muscle was estimated to at 6–8 mmol/L, but decreased to approximately 3 mmol/L during contraction and 1.7 mmol/L when consumption was high
[56]. Experiments using skinned fiber have shown that contractility decreased when the ATP concentration fell below 2 mmol/L
[57]. The concentration of Mg
2+, which was released from MgATP also increased with the consumption of ATP
[58]. However, normal exercise does not substantially reduce the ATP concentration in muscles
[25][59][60], but rather a large amount of ADP is generated as a by-product
[60]. ADP does not maintain RyR activity as efficiently as ATP
[24]. Therefore, the abundant increase in ADP may suppress channel activity. When high load-bearing exercise increases the ADP/ATP ratio, the impact of ADP may be enhanced (
Figure 2B).
The SR luminal Ca
2+ concentration decreased from 1 mmol/L to 0.08 mmol/L after exercise and high load-bearing work in one study
[34]. The RyR senses a reduction in luminal Ca
2+ concentration and channel activity decreases
[30]. In addition, when luminal Ca
2+ is depleted, calsequestrin binds to the RyR and suppresses its activity. Moreover, the acidification of the muscle cytosol by exercise
[25] causes the suppression of RyR activity. Thus, high load-bearing work results in muscle fatigue that suppresses RyR activity (
Figure 2B).
3.2. Skeletal Muscle Injury
Skeletal muscles are close to the body surface and are sensitive to external environments. Therefore, strong pressure or blows can lead to muscle contusions. It is known that muscle injury increases the production of H
2O
2 [61] and NO
[62][63] (
Figure 2C). Although ROS, including H
2O
2 and NO are known to increase RyR activity, an increase in RyR activity does not occur in damaged muscle fibers during exercise because damaged fibers are unable to contract
[62][64]. However, it has been shown that ROS can spread from the site of injury and affect other muscle fibers and tissues. In particular, H
2O
2 has a large impact because it can easily permeate the cell membrane. Excessive contraction of the surrounding fibers because of increased RyR activity may increase damage in the contusion site. It has been reported that cooling of the contusion site reduces ROS production
[64]. Thus, preventing the increase in RyR activity after muscle injury may be an important aspect of early treatment.
3.3. Stresess and Aging
Heat stress and hypoxia at high altitude also affect skeletal muscle. Heat stress is known to cause a transient increase in oxidation
[65], to stimulate mitochondrial ROS production
[66], and to promote the expression of endothelial nitric oxide synthase (NOS)
[67] (
Figure 2C). It has been reported that mice with a malignant hyperthermia mutation in RyR1 display increased RyR activity and rapid fever after the induction of heat stress, with the unfortunate enhancement of mitochondrial ROS production, resulting in sudden death
[68].
Hypoxia at high altitude changes redox state. Under this condition, NOS is induced by the hypoxia-induced transcription factor HIF-1a
[69] and ROS-production increases with anearobic ATP synthesis; therefore, proteins are oxidized
[70] and RyR1 binding by FKBP12 is relieved due to the increased ROS and NO
[71] (
Figure 2C). At high altitudes, muscle injury may be induced by intense contractions because the activity of RyR1 is already increased.
Aging must also be discussed as one of the critical conditions affecting the muscle. Although many studies have been performed on aging and muscle atrophy, there are not many reports on factors that regulate RyR activity. However, it was demonstrated that calsequestrin expression increases with age
[72] (
Figure 2D). Additionally, the coupling of the DHPR and RyR1 is also decreased with aging
[73].
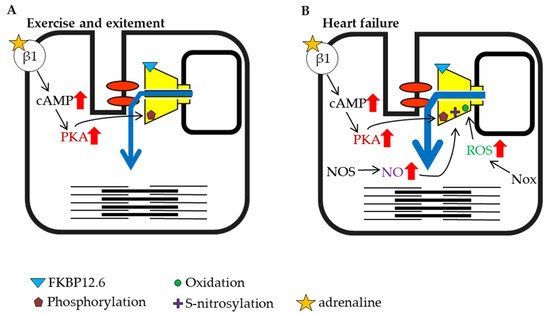
3.4. Modulation of Heart Function in Health and Disease
The heartbeat is caused by regular Ca
2+ release from the RyR2 channel, which is controlled by an action potential originating at the plasma membrane. The rate of the heartbeat is usually 60–100 beats per minute in humans, but it is known to be higher during exercise and psychological stress. When the sympathetic nervous system is stimulated by exercise or excitement, β-adrenergic signaling activates PKA in the myocardium and increases amplitude of cardiac action-potential-induced Ca
2+ release from RyR2 by direct and indirect mechanisms. The factors that enhance RyR2 activity are elevation of SR luminal Ca
2+, which results from an increase in Ca
2+ influx via L-type Ca
2+ channels and enhanced SERCA activity, and increased phosphorylation of RyR2 (S2030 and S2808)
[74][75] (
Figure 3A). NO production is also increased under exercise and stress conditions. These factors all potentiate RyR2 activity and result in strong contractions
[76][77].
Figure 3. Schematic representation of cardiac ryanodine receptor (RyR2) modulation by intracellular factors under different conditions (part 1). (A) Exercise and excitement. (B) Heart failure.
Heart failure is a highly prevalent progressive cardiac disorder with high morbidity and mortality. It arises from various causes, including coronary artery disease leading to cardiac arrest, high blood pressure, atrial fibrillation, valvular heart disease, and cardiomyopathy. When the ejection force is reduced during heart failure, the heart is unable to sufficiently maintain blood flow to meet the oxygen demands of tissues. The sympathetic nervous system is activated in the early stage of heart failure, which promotes PKA phosphorylation of RyR2 and NO production
[78] (
Figure 3B). It was reported that inducible NOS expression increased in failing cardiomyocytes
[79]. In addition to the increase in NOS, an increase in Nox expression has also been reported during heart failure
[80][81]. Nox is an enzyme involved in the generation of ROS
[82][83], and ROS promotes RyR oxidation and contributes to increased channel activity (
Figure 3B). As noted above, phosphorylation, S-nitrosylation, and oxidation may contribute to the activation of RyR2 (
Figure 3B). Moreover, it has been reported that FKBP12.6 dissociates from RyR2 as heart failure progresses
[84][85], which may further contribute to increased RyR2 activity (
Figure 3A). The immediate activation of RyR2 by early compensatory mechanisms that involve these factors may restore cardiac function and ensure the survival of individuals.
The chronic activation of RyR2 has deleterious effects on cardiac function since it causes diastolic Ca
2+ leakage from the SR and generation of abnormal Ca
2+ waves. The former leads to contractile dysfunction and the latter causes arrhythmias. Elevated intracellular calcium levels activate CaMKII, which is also involved in the phosphorylation of RyRs. CaMKII was found to be increased in failing cardiomyocytes
[86] and may be involved in the subsequent development of disease-related ventricular remodeling
[87] or arrhythmia
[88]. Therefore, the excessive activation of RyR2 may lead to the worsening of heart failure rather than improvement of cardiac function. Interestingly, the introduction of calmodulin that had high binding affinity for RyR2 prevented the progression to arrythmia
[89] and reduced the symptoms of CPVT in the animal model
[90]. These results indicate that it may be desirable to return to a moderate level of RyR2 activity in the heart after the initial emergency period.
Similar to heart failure, impaired cardiac function caused by ischemia-reperfusion injury also promotes ROS and NO production, phosphorylation, and dissociation of FKBP12.6 from RyR2
[91]. Ischemia-reperfusion is defined as the restoration of coronary blood flow after an ischemic episode. This injury to the myocardium causes the reduction in ejection force, and the increased demand for oxygen in tissues is similar to that observed for intense exercise and heart failure. It has been reported that the phosphorylation of S2814 by CaMKII is enhanced after ischemia-reperfusion
[92]. Increased oxidation of RyR2 is a result of the increase in ROS generation in cardiomyocytes
[93][94]. The influence of ROS reaches the cardiomyocytes that surround the injured myocytes. Furthermore, ROS produced from tissues other than heart tissue are also involved in RyR2 modulation. Strong oxidative stress responses occur after burns and include the activation of RyR2 and leakage of calcium from the SR in cardiomyocytes, which leads to heart failure
[95].
Heart failure has been shown to have a significant effect on skeletal muscle. Biopsies of skeletal muscle from patients with heart failure have shown that RyR1 binding to FKBP12 is reduced
[96]. It has been reported that phosphorylation of the S2843 residue of RyR1 by PKA dissociates FKBP12 from the receptor, and phosphorylation and dissociation of FKBP12 were also observed in an animal model of heart failure
[97] (
Figure 3B).
 +1 point
+1 point