1. The Effects of Zeolite Structures on Alkane Cracking
The influences of zeolite structures on cracking activity and selectivity are usually attributed to the confinement effects in zeolites, which can be studied by examining the variations in the parameters of alkane adsorption and activation with zeolite topologies
[1][2][3]. In principle, the intrinsic activation enthalpies and entropies (
ΔH‡int and
ΔS‡int) need to be calculated by subtracting the adsorption thermodynamic properties (
ΔH‡ads and
ΔS‡ads) from the apparent activation enthalpies and entropies (
ΔH‡app and
ΔS‡app) derived from kinetic data measured experimentally, so an accurate determination of adsorption enthalpies and entropies is required to determine intrinsic catalytic activities. However, it is difficult to experimentally measure alkane adsorption enthalpies and entropies at cracking conditions (>673 K), so the adsorption properties of alkanes have been evaluated using theoretical calculations
[1][3].
For example, Bell and coworkers demonstrated that
ΔH‡ads and
ΔS‡ads can be calculated accurately using CBMC simulations, which can properly capture the redistribution of adsorbed alkanes to different active sites
[1][3]. Janda et al. calculated the adsorption parameters for propane, butane, pentane, and hexane in H-MFI at the T4 and T9 positions, which are located in the sinusoidal channel and at the channel intersection, respectively
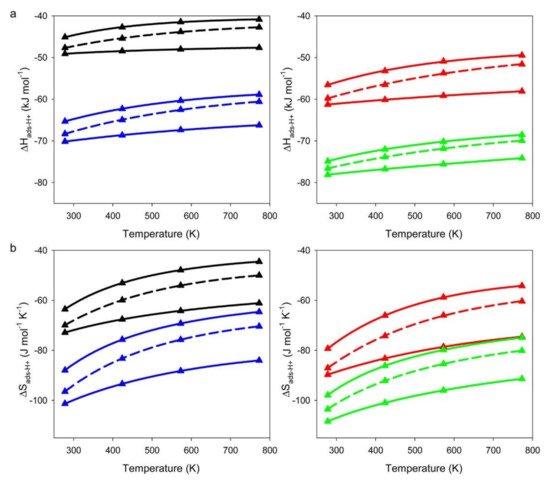
[3]. As shown in
Figure 1, the
ΔH‡ads and
ΔS‡ads are more negative for T4 than for T9, suggesting stronger guest–host interactions between alkanes and the walls of zeolite at the T4 position. This finding suggests that alkanes may prefer to adsorb at the T4 site at low temperatures due to better energy stabilization. On the other hand, at high temperatures, alkanes may prefer to adsorb at the less confined T9 site because of smaller entropy loss for adsorption. However, in practice, the adsorption position is determined not only by temperature but also by the Al distribution, as the distribution of Al in zeolites is often not random and is influenced by the synthesis conditions
[4].
Figure 1. CBMC simulations of (
a) adsorption enthalpies (
ΔH‡ads) and (
b) adsorption entropies (
ΔS‡ads) of propane (black triangles), butane (red triangles), pentane (blue triangles), and hexane (green triangles). The upper and lower solid lines represent acid sites located at T9 and T4, respectively. Boltzmann weighted averages of
ΔH‡ads and
ΔS‡ads for Al distributed evenly between T9 and T4 are illustrated as dashed lines. Reprinted with permission from
[3]. Copyright 2015, American Chemical Society.
Yang et al. investigated the influence of Al distributions on alkane adsorptions in MFI, TON, FER, MWW, MOR, KFI, and FAU with various Si/Al ratios
[5]. They studied the central-to-terminal bond adsorption selectivity ratio of C4-C6 alkanes and found that the influence of Si/Al ratios on the selectivity is negligible. However, their results suggested that the adsorption selectivity ratio is sensitive to Al distributions, because a high selectivity to central bond adsorption was obtained at the T-sites located in a more confined space. In addition, the close spatial proximity of individual Al atoms was also found to enhance the adsorption of central C–C bonds, which indicates that the adsorption selectivity can be tuned by varying the spatial proximity of Al.
Many studies have suggested that the intrinsic activation energy of alkane cracking is not sensitive to zeolite structures
[6][7][8][9][10][11]. For instance, Van Bokhoven and Xu investigated the rate of monomolecular cracking in MFI, MOR, FAU, and BEA
[8], and reported that cracking rates increase with decreasing pore size, but intrinsic activation energies are independent of zeolite structures. Gounder and Iglesia observed similar results when they investigated the intrinsic rate parameters of monomolecular cracking and dehydrogenation of propane in H-MFI, H-FER and H-MOR
[9]. They found that the intrinsic activation barriers were similar for these zeolites, in agreement with the study of propane cracking in H-MFI, H-MOR, H-BEA, and H-FAU by Xu et al.
[6]. This conclusion is also consistent with the calculations of intrinsic activation barriers for monomolecular propane cracking in zeolites with different topologies (FAU, BEA, MOR, MFI, MWW, and FER)
[7]. A similar conclusion was drawn by Kadam et al. when they investigated monomolecular cracking of propane and butane in FER, TON, MFI, and CHA at various temperatures (580–710 K)
[10]. These authors found that the activation energies are insensitive to zeolite structures, though the activation entropies increase significantly with the decrease of the zeolite effective radii, suggesting that the confinement affects the entropy of the adsorbed state more significantly than that of the transition state. Recently, Berger et al. employed a hybrid quantum mechanics:quantum mechanics (QM:QM) method, MP2:(PBE + D2) + ΔCCSD(T), in periodic systems to predict the adsorption and activation energies of propane in H-FER, H-MFI, and H-CHA, and concluded that the intrinsic activation enthalpy is insensitive to zeolite structures
[11]. To summarize, for the zeolite catalysts discussed above, the intrinsic catalytic activity for cracking is structure-insensitive when considering intrinsic activation energies, and the change of apparent kinetics with different pore sizes and structures can be mainly attributed to the adsorption thermodynamics.
Although the studies discussed above suggested that the intrinsic activation energy for cracking was structure-insensitive, recent investigations indicate that the intrinsic kinetics of cracking may differ between different T-sites for some zeolite systems
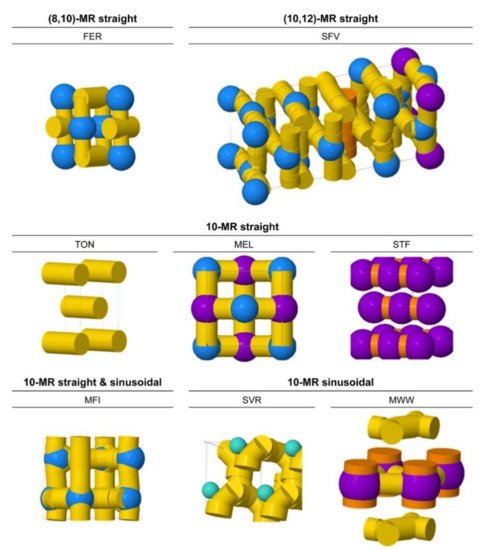
[12][13], which implies that local porous environments can influence not only the thermodynamics of adsorption but also the intrinsic catalytic activity for cracking. To mitigate this gap between different observations, Bell and co-workers investigated the influence of zeolite structures on monomolecular cracking and dehydrogenation kinetics of butane on various zeolites, including TON, FER, SVR, MFI, MEL, STF, and MWW (
Figure 2)
[1]. Using the calculated
ΔS‡ads as a descriptor of the degree of confinement in zeolites, they showed that
ΔH‡int and
ΔS‡int for terminal cracking and dehydrogenation decrease with increasing confinements of acid sites, while
ΔS‡int increases and
ΔH‡int remains nearly constant for central cracking. The variations in the intrinsic activation parameters of different reaction pathways suggest that it is possible to achieve high selectivity for the desired product by carefully choosing a zeolite with proper pore sizes. Recently, the same group used a QM/MM approach to calculate intrinsic and apparent activation parameters, and applied thermal adjustments to the apparent barriers derived from CBMC simulations to account for the changes in activation parameters shown in the experiment
[2]. They investigated butane cracking in three groups of zeolites that feature different cage sizes or channel patterns. Similar to their previous works, they observed that the variations in reaction paths result in different trends of activation parameters with zeolite topologies. This finding does not support the conclusion drawn by previous studies that intrinsic activation parameters are structure-insensitive and pore topologies only affect adsorption thermodynamics
[6][7][8][9][10][11]. The discrepancy of the conclusions drawn by different works leaves the effect of local porous environments for intrinsic cracking activity an open question requiring further investigation.
Figure 2. Characterizations of 10-membered ring (MR) zeolites with different channel and cavity topologies (generated with the ZEOMICS web tool
[14]). Channels are represented in yellow (<6 Å diameter) and orange (>6 Å diameter), while cages are illustrated as green (<6 Å diameter), blue (6–8 Å diameter), and purple (>8 Å diameter) spheres. Reprinted with permission from
[1]. Copyright 2016, American Chemical Society.
2. The Effects of Mesoporosity on Alkane Cracking
Zeolites have been widely used in the petrochemical industry due to the advantage of excellent hydrothermal stability, strong Brønsted acidity, and shape selectivity. However, microporous zeolites with long diffusion paths experience strong diffusion limitation. This problem manifests itself in two scenarios. The first is that large molecules may not be able to diffuse through the channels to react with an active site. The second is that the cracking products would be stuck in the micropores due to diffuse limitation, and further be over-cracked into undesired byproducts, which could lead to coke formation and the subsequent deactivation of the catalyst.
One way to address this problem is to implement larger pores in zeolites
[15][16][17][18][19]. Previous works have successfully fabricated hierarchical zeolites with mesopores of 2–50 nm in diameter with high hydrothermal stability, strong acidity, and low diffusion limitation
[17]. In general, mesoporosity can be created using either the top-down or bottom-up methods. The top-down approach is to remove silica or aluminum from the zeolite framework via dealumination or desilication. The drawback of this approach is that it may suffer material loss and it is difficult to control mesoporosity using the top-down approach
[17]. These issues can be avoided by using bottom-up methods, which use templates to generate ordered mesoporous structures in the synthesis process. However, the bottom-up method is often more costly than the top-down dealumination/desilication method, and therefore may not be economically feasible for large-scale production
[17]. Noteworthy is that the surfactant-templated top-down method can introduce uniformly distributed mesopores into zeolites, while maintaining the high hydrothermal stability and strong Brønsted acidity, and does not suffer from the drawbacks of material loss and damage of catalyst
[15]. In comparison to conventional zeolites, zeolites with secondary porosity display better catalytic performance and longer catalyst lifetime. For instance, mesoporous zeolite Y has been tested in the cracking of vacuum gas oil (VGO). The result showed mesoporous Y produced less coke and more gasoline and light cycle oil (LCO) compared to conventional Y zeolite.
Molecule dynamics (MD) simulations have been applied to study the diffusion of molecules in zeolites at different operation conditions. For instance, Bail et al. investigated the diffusion of hexane in hierarchical mesoporous H-ZSM-5 from 363 K to 543 K using MD simulation and their results indicated that the diffusivity of hexane in a mesostructured zeolite can be higher than in a conventional zeolite at high loading or elevated temperatures
[20]. However, diffusing along the mesopore surface at low loading leads to more tortuous diffusion paths, which may cause a reduction in diffusivity. Bu et al.
[21] when they investigated the diffusivities of coke precursors such as benzene, naphthalene, and anthracene in mesoporous H-ZSM-5 using MD simulations
[22]. They found that these molecules mainly diffuse through micropores or along the external surface of mesoporous zeolites at low temperature, while diffusing along mesopores at pyrolysis temperatures. Recently, Josephson et al. used Monte Carlo simulations to investigate the adsorptions of furan, hexanoic acid, hexane, decane, tetradecane, and 3,6-diethyloctane in hierarchical mesoporous ZSM-5
[23]. Their simulations demonstrated that as pressure increases, alkanes first fill the micropores, then the surface of mesopores and finally the interior of mesopores. In contrast, furan and hexanoic acid predominately adsorb on the surface of mesopores because of hydrogen bonding interactions between adsorbates and silanol groups. These works demonstrated that various guest–host interactions can affect adsorption sites and diffusion paths in zeolite pores, and hence could potentially influence the overall performance of a zeolite catalyst.
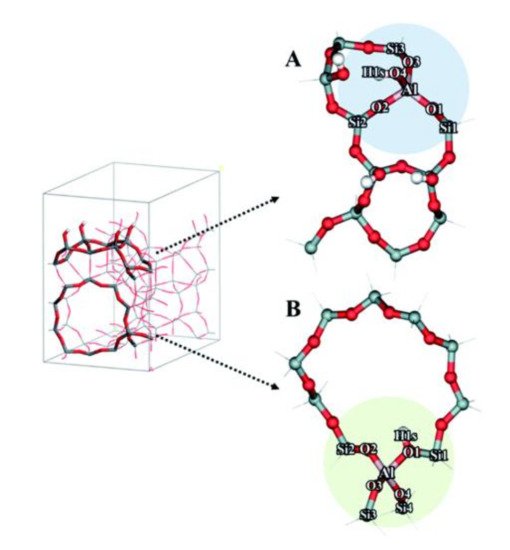
As discussed above, DFT studies have provided valuable insights to zeolite catalysis. However, applying DFT calculations to investigate reactions in mesoporous zeolites is very challenging due to the increased structural complexity. With the presence of both microporous and mesoporous channels, the local environment of active sites can vary dramatically from one position to another, so the number of model systems and reaction pathways that need to be considered for a hierarchical zeolite is often higher than that for a conventional zeolite. For example, to study the effect of hierarchical surfaces on ethanol dehydration, Shetsiri et al. built two different cluster models to represent hierarchical and conventional H-ZSM-5 (shown in
Figure 3), and found that the preferred dehydration mechanism is dependent on the model system used
[24]. Based on both experiments and DFT calculations, these authors demonstrated that direct dehydration of ethanol to form ethylene possibly occurs over the active site at the external surface of hierarchical H-MFI (
Figure 3a), while the dehydration of two ethanol molecules to form diethyl ether is preferred for the internal acid site of H-MFI (
Figure 3b). This finding highlights the importance of incorporating a significant part of the zeolite structure into the model. Although the use of a simplified model can reduce computational costs, a small or medium model cannot fully describe the effects of confinement and dispersion effects in porous structures
[25], which can significantly influence reaction kinetics in zeolites. Recent developments in quantum mechanics/molecular mechanics simulations offer an efficient way to solve this problem
[26]. In the QM/MM hybrid scheme, only a small cluster encompassing the active center is described by quantum mechanics, while the rest of the zeolite is described by a classical force field. The QM/MM scheme allows the simulation of reactions in porous environments without significantly increasing computational costs
[27][28][29], and hence may be used increasingly to model complex mesoporous zeolites in the future.
Figure 3. The optimized cluster models of (
A) hierarchical H-ZSM-5 and (
B) conventional H-ZSM-5. Reproduced from Ref.
[24] with permission from the Royal Society of Chemistry.
 +1 point
+1 point