Ectosolic 5′-nucleotidase (CD73) (EC 3.1.3.5) is a 70-kDa glycosylated protein bound to the outer surface of the plasma membrane by a glycosylphosphatidylinositol anchor. The enzyme is overexpressed in a variety of tumours
[5] and in some of them, is associated with a highly invasive cancer phenotype, drug resistance, and tumour-promoting functions
[6]. CD73 catalyses the dephosphorylation of extracellular AMP to adenosine (Ado), which plays important roles in many physiological and pathophysiological conditions through G-protein coupled Ado receptors (A1, A2A, A2B, and A3)
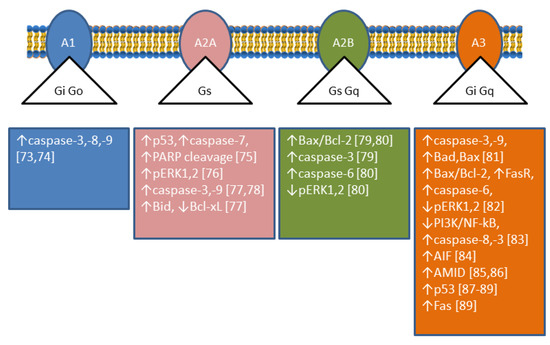
[7][8]. The roles played by Ado are complex since its interaction with different receptors achieves different results; in particular, A1 and A3 receptors are coupled with Gi-proteins determining a decrease in cyclic AMP (cAMP), while A2A and A2B receptors are coupled with Gs-proteins causing an increase in intracellular cAMP
[9] (
Figure 3). Therefore, the role played by CD73 depends on the nature and distribution of the Ado receptors in a particular kind of cell. In addition, CD73 plays also many different roles in cell physiology not related to its catalytic activity
[10]. Analysing enzymatic and non-enzymatic functions of CD73, it was concluded that both faces were involved in the aggressive behaviour of cancer cells. The enzymatic function seems to be primarily involved in invasion, whereas the non-enzymatic action of the protein contributes to cell adhesion and migration through activation of focal adhesion kinase
[10]. Among the tumours in which CD73 is upregulated, breast cancer is the most studied
[11]. In some cases of this type of cancer, an involvement of CD73 enzymatic activity was demonstrated, since supply of Ado could be a substitute for enzyme upregulation in promoting proliferation and motility
[12]. In other cases, the involvement of different not yet described mechanisms independent of enzyme activity was demonstrated
[13]. In all cases, an activation of the Akt/GSK-3β pathway was found to be involved in the tumour growth and motility promoted by CD73
[13]. In human cervical cancer cells, an increase in cell proliferation and motility was associated to CD73 overexpression, but the mechanism was independent of enzyme activity. In fact, CD73 inhibitors were unable to prevent the increase in proliferation in cells that overexpressed the enzyme. In addition, an increase in Ado, that could be expected when the enzyme is overexpressed, induced a decreased cell proliferation
[14]. In this type of tumour, an activation of the Akt pathway was demonstrated as well
[14]. In colorectal cancer, a downregulation of miR-30a was shown to determine an increase of CD73 expression in tumour cells, which promoted proliferation and inhibited apoptosis. MiR-30a is one of the most important tumour-suppressor factors in various human cancers and its level is significantly decreased in several tumours
[15]. Since, as stated before, many effects exerted by the expression of CD73 on tumours are mediated by the participation of the enzyme activity in the conversion of extracellular ATP into Ado, such effects are dependent on the amount and nature of the Ado receptors expressed by tumour cells and other cells present in the tumour microenvironment. In this regard, in contrast to reports for other cancer types (see above), in an in vivo study on medulloblastoma, overexpression of CD73 reduced tumour growth and vascularization, and also promoted differentiation and initiated apoptosis, supposedly by the accumulation of Ado which interacted with A1 receptor
[16]. The level of CD73 expression is very important not only for tumour growth and motility, but also for the success of the therapeutic approach. In fact, during chemotherapy, various immunogenic mediators accumulate in the tumour microenvironment, included ATP. Extracellular ATP, in this case, is released by the cells undergoing intrinsically or extrinsically activated apoptosis, through pannexin-1 channels and functions as a “find me” signal for P2Y family receptors expressed by macrophages and dendritic cells
[17]. In this mechanism, the rapid ATP degradation catalysed by several extracellular enzymes is a determinant for the immunogenic activation, since the accumulated AMP, which cannot interact with P2Y receptors, can generate Ado through CD73, which mainly mediates the immune-escape of tumour cells interacting with A2A receptors
[18]. Moreover, also in the case of regulatory T-cells (Tregs) in a tumour environment, the presence of several signals triggers apoptosis, and apoptotic Treg cells achieved superior immuno-suppression via an oxidative stress-associated mechanism
[19]. Therefore, the induction of apoptosis in a tumour environment in which cells highly expressing CD73 and Ado receptors A2A are present is not always a successful therapeutic approach. Recently, CD73 has been targeted for the synthesis of new inhibitory compounds which prevent extracellular Ado formation from AMP and the consequent immune-escape
[20].
 +1 point
+1 point