1000/1000
Hot
Most Recent
 +1 point
+1 point
Neurodegenerative diseases (NDDs) are becoming increasingly prevalent in an age-dependent manner, partly because life expectancy has increased in recent years due to our advanced medical knowledge. The most prevalent of NDDs includes Alzheimer’s disease (AD), Parkinson’s disease (PD), Huntington’s disease (HD), amyotrophic lateral sclerosis (ALS), frontotemporal dementia (FTD), spinocerebellar ataxias (SCA), prion diseases (PrD) and others.
Another common feature underlying neurodegeneration is the formation of stress granules (SGs). SGs are subtypes of RNA granules that assemble from the interaction of RNA-binding proteins (RBPs) with untranslated messenger ribonucleoproteins (mRNPs). These mRNPs are formed from mRNAs halted in translation initiation due to stress response or drugs [1]. SG components that do not bind RNA are presumably recruited to SGs through protein-protein interactions. These RNA–protein or protein–protein complexes form membraneless organelles that usually occur via liquid–liquid phase separation (LLPS). The formation of these membraneless organelles is a strategy of cellular compartmentalization that plays a role in several fundamental physiological processes. Interestingly, many RBPs contain “low complexity domains” (LCDs), also referred to as intrinsically disordered protein regions (IDPRs), which consist of different residues that facilitate binding affinity between SG components. The binding between SG components may result in undesired amyloid aggregation formation [2][3] and subsequent neurodegenerative responses [4]. The proteins best characterized in this regard include hnRNPA1, hnRNPA2, Fused in Sarcoma (FUS), TAR DNA-binding protein 43 (TDP43) and tau [5]. The nuclear pore complex (NPC) is another target in the cell that interfaces the formation of pathological SGs. Under stress, nuclear pore components, 30 types of proteins called nucleoporins (NUPs), and other proteins of the nucleocytoplasmic transport (NCT) system translocate to the cytoplasm and are sequestered to existing SG [6].
Impairment of the NPC, in general, and NCT, in particular, has recently emerged as a central disease mechanism in different NDDs ( Figure 1 ). Proteins that are smaller than 40 kDa can diffuse freely across the nuclear membrane; however, proteins above 40 kDa require active transport to cross the nuclear membrane. Thus, NCT refers to the active import and export of large molecules from the cell nucleus via the NPC. This process facilitates the transition of a protein possessing either a nuclear localization sequence (NLS) and/or nuclear export signal (NES) which are recognized by specific auxiliary carrier proteins, importins, and/or exportins, respectively. Dating back to 2015, a connection was revealed between the disruption of NPC components as well as the nuclear import–export machinery and different models of the chromosome 9 open reading frame 72 (C9ORF72) ALS-linked mutation [7]. Following this publication, extensive research progressed in this regard, with consistent results substantiating the disruption of intact NCT in ALS, caused by different mutations: TDP43 [8], FUS [9], PFN1 [10], and in other NDDs, including AD [11] and HD [12][13].
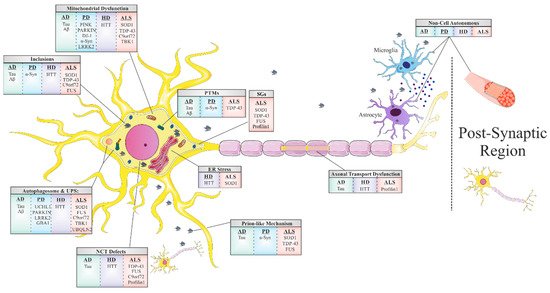
The majority of adult NDDs are characterized by intra- or extracellular aggregation of misfolded proteins, a subject that will be further discussed later in this review. These misfolded proteins, frequently, possess self-propagation properties as a mechanism of spreading in an individual organism, another common feature of NDDs often referred to as a ‘prion-like’ mechanism ( Figure 1 ). In the self-propagation mechanism, the normal form of the prion protein (PrPC) undergoes a conformational change into a misfolded protein, by interacting with pathogenic prion protein (PrPSc). The ability of misfolded proteins to ‘seed’, that is, to recruit physiological proteins of the same kind and to induce their conversion into a pathological form, and propagate from cell-to-cell, with the continuous conversion of the normal protein into its misfolded form, is eventually the cause of the formation of amyloid aggregates [14]. In vitro and in vivo studies indicate that amyloid beta (Aβ) and tau in AD, alpha-synuclein (α-syn) in PD [15], and TDP43 and SOD1 in ALS [16] have similar “prion-like” characteristics. Interestingly, prion-like diseases arise not only through inherited mutations in the prion protein but also sporadically from the wild-type form of the protein [17].
The last and most remarkable common mechanism of NDDs is the misfolding event of key proteins that accumulate and form toxic aggregates which disrupt the proper functioning of the cell’s different processes ([18] and Table 1 ). This well-known feature is the foundation of all other mechanisms we have mentioned before in this review, and it has a large impact on our understanding of these mechanisms. Fascinating research from genetic, neuropathological, cellular, and biochemical studies, as well as from experiments with in vivo models [19] and postmortem tissues [20], confirm that protein misfolding, oligomerization, and accumulation in the CNS are the main events triggering pathological abnormalities that are responsible for NDDs. These proteins undergo misfolding from their native states to form β -sheet-rich structures, ranging from small oligomers to large fibrillar aggregates. For each NDD, there is one or more prototype protein which is known to misfold, accumulate, and form aggregates. These proteins include Aβ and tau in AD; α-syn in PD, TDP43, SOD1, C9Orf72, and FUS in ALS; TDP43 and tau in FTD; and Huntingtin (HTT) in HD. In healthy cells, misfolded proteins are either degraded or refolded correctly by chaperones (proteins that are involved in protein folding). However, if the misfolded protein accumulates into an amyloid aggregate with a fibril structure, then it has a higher resistance to degradation due to its extremely stable thermodynamic state. This state enables it to convert more native proteins into an amyloid form (as mentioned in the ‘prion-like’ mechanism), thereby escalating the pathology of the cell, leading to the progression of the disease [21].
| Disease | Associated Genes | Associated/Pathogenic Protein | Normal Function |
Toxicity Mechanism | Related Chaperones | Current Treatments |
|---|---|---|---|---|---|---|
| AD | MAPT | Tau | Polymerizes tubulin into microtubules | Gain and loss of function | HSP90 HSP104 HSP70 HSP60 HSC70 |
AChE inhibitors:
|
| APP PSEN1 PSEN2 |
Amyloid β | There are evidence for its precursor participation in:
|
Gain of toxic function | |||
| PD | SNCA | α-Synuclein |
|
Gain of toxic function | HSP70 HSP40 HSP90 |
Precursor of dopamine
|
| UCHL1 | UCHL1 | Neuron-specific deubiquitinating enzyme | Gain of toxic function | |||
| PARK2 | Parkin |
|
Loss of function | |||
| PINK1 | PINK1 | function of the mitochondria and mitophagy function of the mitochondria and mitophagy
|
Loss of function | |||
| PARK7 | DJ-1 |
|
Loss of function | |||
| LRRK2 | LRRK2 | Mitochondrial clearance | Gain of toxic function | |||
| GBA | GCase | Lysosomal enzyme | Gain and loss of function | |||
| HD | HTT | HTT |
|
Gain of toxic function | HSP70 HSP40 HSP90 |
Only supportive treatments are available |
| ALS | SOD1 | SOD1 | Catalyzes the conversion of superoxide radical into peroxide and oxygen | Gain of toxic function | CCS HSP70 HSP40 HSP110 HSPB8 HSP27 MIF 4-PBA |
Glutamate antagonist:
|
| TARDBP | TDP-43 | RNA metabolism | Gain and loss of function | |||
| C9orf72 | C9orf72 | Yet to be established | Gain and loss of function | |||
| FUS | FUS |
|
Gain and loss of function | |||
| UBQLN2 | Ubiquilin2 | Degradation of ubiquitinated proteins | Yet to be established | |||
| TBK1 | TBK1 |
|
Loss of function | |||
| PFN1 | Profilin1 | Actin polymerization | Gain and loss of function |
Beyond the role of proteins and their conformational changes in the process of neurodegeneration, they are an essential component in the biological system as a whole, facilitating almost every process, and thus, their correct formation and regulation is crucial for intact cellular function. The requirement of each protein differs within the cell and varies among diverse cells and demands; therefore, there is a need for a strict system to manage the proteins homeostasis.
The folded structure of proteins must retain conformational flexibility to function; thus, a compromise exists between thermodynamic stability and conformational stability. They are only marginally thermodynamically stable in their physiological environment [22], and thus, along with physiological stress conditions such as heat, oxidative stress, and inflammation, proteins are susceptible to the formation of non-native interactions that may lead to protein misfolding and aggregation [23].
The protein-coding sequence contains all the needed information to achieve the correct final structure of the protein; however, this process is challenged due to timescale constraints and protein overload [22]. Protein overload, which is characterized by the presence of a large amount of different proteins, enhances protein aggregation by increasing the affinities between the interacting macromolecules, including folding intermediates. Furthermore, large proteins with complex structures may expose hydrophobic amino acid residues to the solvent during their folding, leaving them susceptible to non-native interaction that might lead to aggregation [24]. Although aggregation primarily leads to amorphous structures, it may lead to the formation of fibril-like amyloid aggregates [24], which are more toxic to the cells, as previously described [25][26]. As a means to facilitate correct protein folding, there is a need for molecular chaperones. These chaperones bind to the exposed hydrophobic residues to shield them from aggregation and allow the protein to fold natively [23].
Besides the chaperones’ fundamental role in de novo protein folding, they are also involved in various aspects of proteome maintenance, such as macromolecule complex assembly, protein transport, protein degradation, aggregate dissociation, and refolding of stress denatured proteins [24].
Alzheimer’s disease (AD) is the most common form of elderly age dementia [27], with an increasing prevalence with age, ranging from 3% at the age of 60 to 32% at the age of 85 and older [28]. It has a higher occurrence rate in females than in males and is considered a common cause of death in elderly individuals [29]. As life expectancy increases, the prevalence of AD and other dementias will increase accordingly [28].
Furthermore, pathological tau can directly interact with components of the NPC that can accelerate aggregation and fibrilization of tau in the cytoplasm and disrupts NPC structure and function. Full-length tau was shown to interact with NUP98 and to a lesser extent with other phenylalanine-glycine containing NUPs, in postmortem AD, in transgenic mouse models, and In vitro. In addition, evidence exists for NPC structural defects, including NUP98 pathology, and functional impairments, including Ran mislocalization, in phospho-tau-positive cells from AD patients, rTg4510 transgenic mice, and primary neurons [11].
Another Aβ clearance pathway is perivascular drainage, which is impaired in AD [30]. Known factors affecting perivascular drainage of Aβ include ApoEε4, deposition of immune complexes, and arterial age. The presence of ApoEε4 is associated with reduced perivascular drainage of Aβ, by competing with Aβ for its efflux by Low density lipoprotein receptor-related protein 1 (LRP1), a receptor from the LDL receptor family, from the interstitium to the circulation. ApoE has three major isoforms (ApoEε4, ApoEε3, ApoEε2), of which ApoEε4 is the strongest risk factor for AD since it is the least efficient at mediating Aβ clearance than are the other ApoE isoforms [31].
Currently, there are four FDA approved treatments available for AD patients, all of which provide only limited therapeutic benefits. The treatments target AD neuropathology which includes elevated glutamate levels in the cerebral spinal fluid. High glutamate levels are believed to disrupt cellular communication and contribute to neuronal loss including loss of basal forebrain cholinergic neurons, leading to a decreased availability of acetylcholine at the neuronal synapse contributing to memory decline [32]. Three of the approved treatments (Donepezil, Rivastigmine, Galantamine) are acetylcholinesterase inhibitors that increase the availability of acetylcholine at synapses and improve cholinergic transmission. These drugs were shown to maintain mental functions by improving cognition, daily and global function, and some behavioral manifestations of AD. The last approved treatment is Memantine, an N-methyl-D-aspartate (NMDA) receptor antagonist that blocks the effects of sustained, pathologically elevated levels of glutamate in order to decrease the neuronal dysfunction and the excitotoxicity injury to the brain. However, the efficacy of Memantine administration in patients with AD remains inconclusive [33].
Although α-syn plays a fundamental role in PD, it seems that loss of protein function is not the only contributing factor to the development and progression of the neurodegeneration observed in PD, as α-syn knockout (KO) mice did not show any symptoms or sign of neurodegeneration. However, these results can be explained by functional redundancy between α-syn and the closely related β- and γ-synuclein. Therefore, a triple KO to all synuclein proteins was made causing some behavioral abnormalities and alterations in synaptic neurotransmission. However, no signs of neurodegeneration were observed [34]. Thus, it is more likely that α-syn mediates neurodegeneration in PD via a gain of a new toxic function.
The emerging of synucleinopathies is thought to be through the process of soluble α-syn conversion into insoluble aggregates via spontaneous nucleation [35]. In 2003, Braak and colleagues, using histopathological studies in post-mortem PD patients, described their hypothesis of the retrograde transport of α-syn from the gastrointestinal tract via the vagus nerve to the ventral midbrain, where it selectively degenerates dopaminergic neurons of the substantia nigra (SN) [36], later supported by mouse model studies [37]. Moreover, the Braak group presented an association between α-syn pathology in different brain regions and PD patients’ symptoms [38]. Their observations provide support for the prion-like mechanism attributed to α-syn, as a cause for PD, which was suggested before [39]. In accordance, heavy metals such as copper and iron, which are known to be accumulated in PD brains [40][41], were found to accelerate prion-like propagation of α-syn fibrils [42].
Parkin is a protein encoded by the PARK2 gene consisting of 465 amino acids. Parkin protein contains two RING (Really Interesting New Gene) finger domains separated at the C terminus by an in-between-RING (IBR) finger domain and a ubiquitin-like (Ubl) homologous domain at the N terminus. IBR presence led to the finding that parkin is an E3 ubiquitin ligase [43]. There is a handful of research about the substrates of parkin that might play role in cell death [44], and some of these substrates seem to link parkin and α-syn [45]. Disease-causing mutations in parkin lead to loss of normal protein function by different mechanisms including exon deletions, missense, and nonsense mutations [46][47][48]. However, impairment of parkin protein function can be due to nitrosative stress, dopaminergic stress, and oxidative stress, which are key pathogenic processes of sporadic PD [49].
In 2002, the importance of chaperones in PD was first revealed when overexpression of HSP70 diminished neurodegeneration mediated by α-syn in a drosophila model. Moreover, overexpression of HSP70 and HSP40 members enhanced degradation of misfolded α-syn, reduced α-syn oligomer formation and toxicity [50][51]. Accordingly, upregulating HSP70 may also prevent the release of extracellular α-syn and thus prevent or slow the propagation of α-syn pathology and disease progression [52]. Interestingly, inhibition of HSP90 chaperone activity results in activation of heat shock factor-1 (HSF-1) and subsequent activation of protective stress-induced HSPs such as HSP70 [53]. Hence, upregulating HSP70, either directly by overexpression or indirectly via inhibition of HSP90, could be a favorable therapeutic approach to PD. Furthermore, HSP90 was found to interact with α-syn and promote fibril maturation in an ATP-dependent manner [54], supporting the beneficial effect caused by the inhibition of these proteins. In addition, overexpression of HSPA5, one of the HSP70 family members, reduces aggregation of α-syn by a refolding activity and by protein degradation through the UPS [55]. Finally, because HSPs have emerged as potentially therapeutic modifiers of cytotoxicity in PD, various drugs are currently in clinical trials in order to target chaperones which show promising effects in PD.





