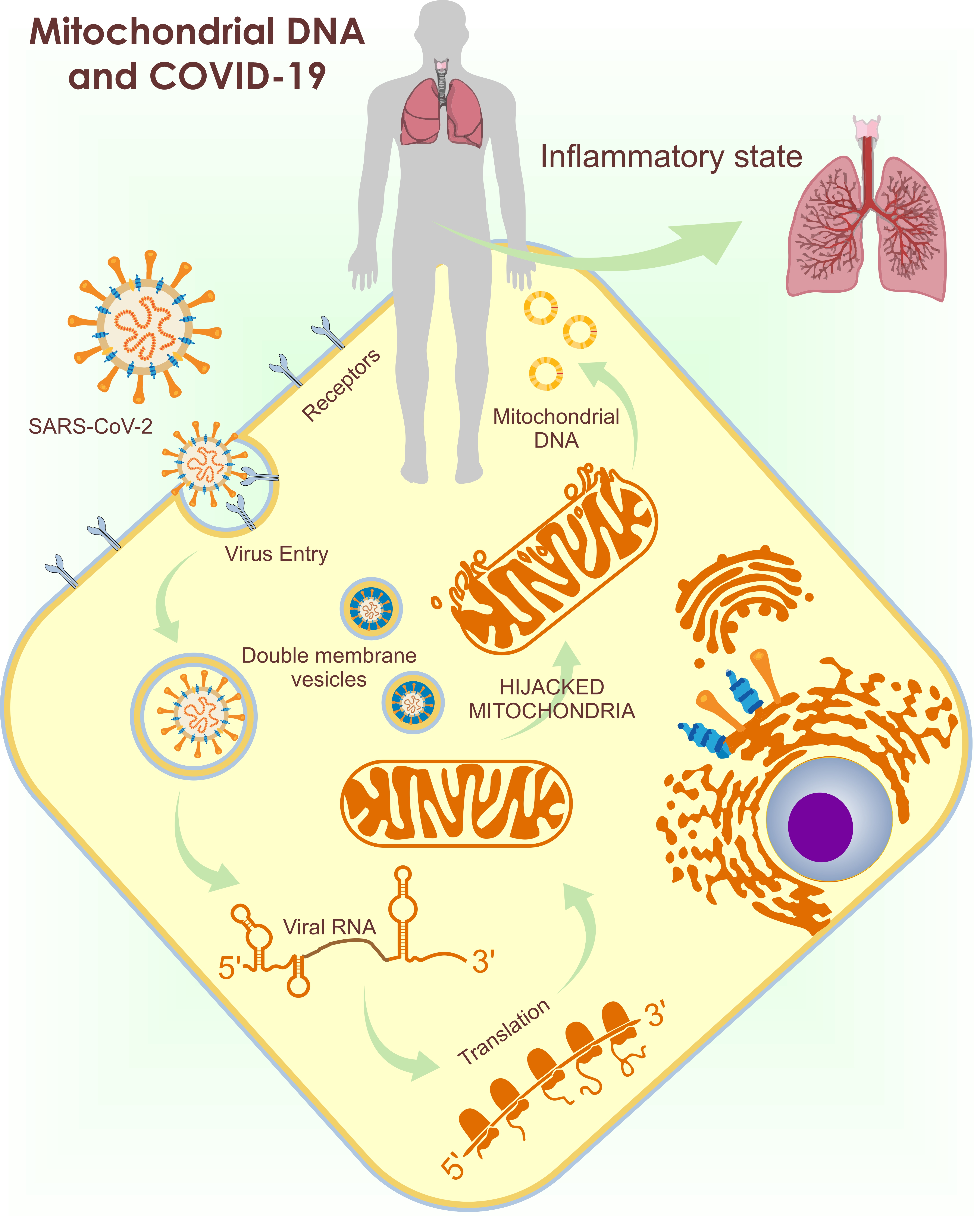
The importance of mitochondria in inflammatory pathologies, besides providing energy, is associated with the release of mitochondrial damage products, such as mitochondrial DNA (mt-DNA), which may perpetuate inflammation. It should be noted the importance of mitochondria, as organelles that produce energy and intervene in multiple pathologies, focusing mainly in COVID-19 and using multiple molecular mechanisms that allow for the replication and maintenance of the viral genome, leading to the exacerbation and spread of the inflammatory response.
 1. Introduction
1. Introduction
Coronavirus disease 19, (COVID-19) is a disease caused by the infection of the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), which was identified for the first time in the city of Wuhan, in the Hubei Province, China in December 2019, causing a global epidemic that has presented a significant threat to the health of the world’s population. As of September 2021, there have been approximately 218,921,481 confirmed cases of COVID-19 around the world and approximately 4,549,975 deaths
[1].
SARS-CoV-2 belongs to the beta subgroup of coronaviruses. This virus measures approximately 120–160 nm in diameter, with a genome made up of a positively charged RNA
[2] and a petal-shaped projection, called the spike (S) protein. This S protein mediates virus binding and membrane fusion during the infection
[3].
SARS-CoV-2 enters host cells by the mechanism of viral spike protein binding to the surface receptor for angiotensin-converting enzyme 2 (ACE-2), which mostly allows for the entry of the virus into type II pneumocytes in the lung of the host
[4]. This induces a local inflammatory process and promotes the release of multiple cytokines, such as tumor necrosis factor alpha (TNF-a) or interleukin-1 beta (IL-1B) and IL-6, which recruit circulatory leukocytes and amplify the systemic inflammation
[5].
The biochemical and pathological features produced by COVID-19 leads to a state of acute inflammation, which is correlated with subsequent destructive effects, manifested as persisting hypoxia, hypercoagulability, acidosis, and altered aerobic glycolytic metabolism, with an elevated lactate dehydrogenase (LDH) and subsequently lactic acidosis
[6]. This inflammatory condition could be related to hypermetabolic states, such as hyperglycemia, and with cellular alterations, such as mitochondrial dysfunction, due to its various functions in metabolic pathways and cellular functions
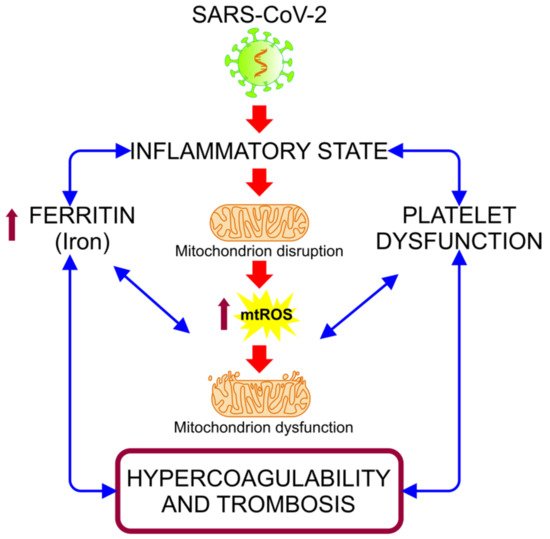
[7][8]. The hyper-inflammatory state associated with COVID-19 induces oxidative stress (OS) events, alterations in iron homeostasis, and states of hypercoagulability and thrombosis (
Figure 1). These events are closely related to mitochondria, organelles that have received minor attention in connection with the pathogenesis of COVID-19, despite being essential in organs and cells with a high metabolism that are highly affected in this disease, such as hepatocytes, pneumocytes, endothelial cells, cardiomyocytes, renal cells, and neurons
[7].
Figure 1. Alteration of iron homeostasis and states of hypercoagulability and thrombosis caused by the hyper-inflammatory state associated with COVID-19. The relationship between the inflammation caused by SARS-CoV-2 infection and the increased ferritin and platelet dysfunction generates a vicious cycle, in which the mitochondrial dysfunction intensifies, inducing an increase of the mitochondrial reactive oxygen species (mt-ROS) and causing an exacerbation of this cycle, resulting in a state of hypercoagulability and thrombosis
[9].
2. The Mitochondrial Role in SARS-CoV-2 Replication
As previously mentioned, SARS-CoV-2 uses ACE-2 to enter the cell
[10]. Once in the cytoplasm, SARS-CoV-2 needs to initiate replication from a single-stranded RNA (ssRNA) through an intermediate dsRNA
[11]. This process predisposes the virus to detection by TLRs and mitochondrial viral signaling systems (AVMs)
[7][8][12][13][14]. SARS-CoV-2 evades this detection by inducing the production of double-membrane vesicles with the help of the mitochondria and the ER for their replication and dissemination
[12]. These vesicles, in addition to being an appropriate site for replication, trick the host into not digesting them. The importance of the mitochondria for the virus replication explains the presence of the SARS-CoV-2 RNA genome and all the produced sub-genomic RNAs in the host MM and nucleolus
[14][15].
Based on these models and in the identification of an open reading frame (ORF), the SARS-CoV-2 viral genome has been identified in mitochondria, indicating that mitochondrial residency could be required for double-membrane vesicle formation, which is critical for the unstoppable replication of coronavirus, as it evades cellular defense
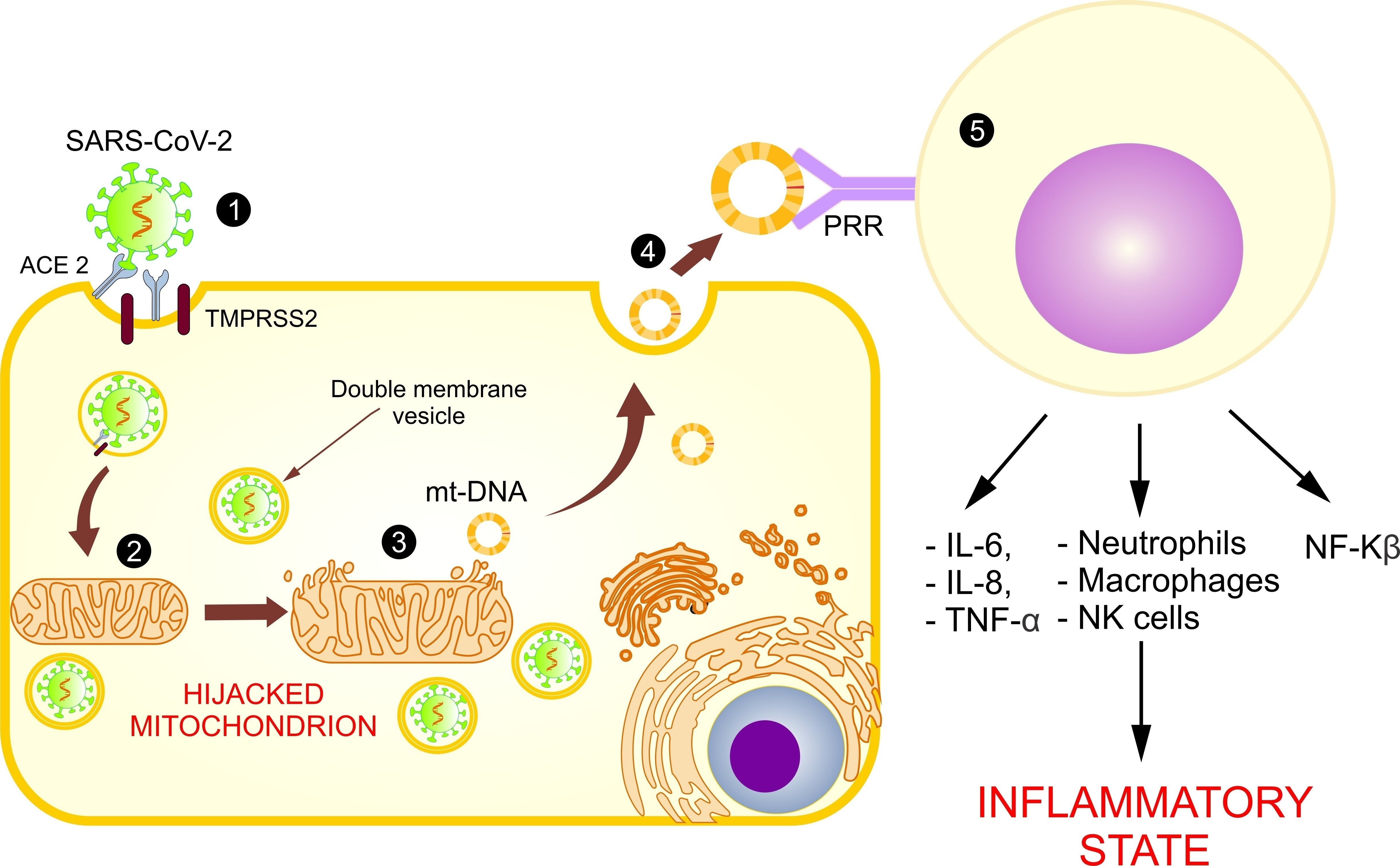
[7]. With these findings, it is assumed that the coronavirus hijacks the mitochondria and uses their machinery for its own sustenance and replication, and the mitochondria eventually assist with its virulence and transmissibility (
Figure 2). These actions may damage the mt-DNA and mitochondrial membrane, causing leakage of the altered mt-DNA into the cytoplasm, and consequently act as a trigger for innate immunity activation.
Figure 2. Use of mitochondria for SARS-CoV-2 replication and the release of mt-DNA into the circulation. The figure shows the introduction of SARS-CoV-2 into the host cell (1) and the hijacking of the mitochondrion for its replication (2), causing a loss of its membrane and the release of mt-DNA into the cytosol (3) and circulation (4), activating the immune system (5), and increasing the inflammatory state.
3. Mitochondrial Dysfunction in COVID-19
Most of the patients infected with SARS-CoV-2 showed either a mild infection, with no fever nor pneumonia, or a moderate infection, with clinical manifestations, such as cough, fever ≥ 38 °C, arthralgias, myalgias, and dyspnea
[16]. Nonetheless, a severe infection caused pneumonia and respiratory failure, accompanied by other complications, such as microvascular thrombosis, coagulopathy, ARDS, disruption in iron homeostasis, and shock
[17]. While the factors that interfere in the infection’s severity are not fully understood, the immune system and mitochondrial function might play an important role in the pathogenesis of the disease.
3.1. Mitochondria and Cytokine Storms
The hyper-inflammatory state caused by COVID-19 in the fight against SARS-CoV-2 infection is a hallmark of the release and amplification of a cytokine storm identified in patients with severe COVID-19, which is regulated by multiple mitochondrial pathways. A cytokine storm occurs due to an extensive increase in ROS, which results in the release of TNF-a, IL-1β, IL-6, and IL-18, which mediate and intensify the inflammatory state and promote the inflammasome response. As a consequence of the activation of a cytokine storm, the cells reprogram their metabolic machinery by increasing the glycolysis and reducing the Krebs cycle’s ATP production, thus inducing mitochondrial atrophy
[18]. Further, inflammatory cytokines, such as TNF-a, IL-6, IL-10, and IFN-γ, are involved in the exacerbation of the inflammatory state and in mitochondrial dysfunction. TNF-a induces calcium-dependent increases in mt-ROS. Moreover, IFN-γ has been shown to upregulate genes, thus inducing mt-ROS generation and increasing intracellular OS. Finally, IL-10 and IL-6 were found to modulate mt-ROS generation by regulating the activity of the ETC
[19]. These inflammatory and immune sentinels trigger intracellular cascades that alter mitochondrial metabolism and increase its dysfunction. Cytokines, such as IL-6 and TNF-a, found in the serum of patients with COVID-19, impede the mitochondrial OXPHOS pathway, causing a decrease in the ATP production and an abnormal mt-ROS production
[20], which support cellular diseases and ageing. This causes mitochondrial membrane permeabilization, the release of mt-DNA, and altered mitochondrial dynamics, and it could ultimately result in cell death. It has also been demonstrated that the differential gene expression in SARS-CoV-2-infected lung cell lines shows an upregulation of the genes involved in mitochondrial inflammatory/cytokine signaling, causing a downregulation in genes involved in the mitochondrial functions of respiration, organization, and autophagy
[21]. As an example, the study conducted by Zhang et al. in 2020 revealed that alveolar epithelial cells in humans with dysfunctional mitochondria show an increased production of some pro-inflammatory cytokines, such as IL-6, IL-12, chemokine ligand 8 (CXCL-8), CCL-20, CCL-3, and CCL-4, all of which were found to be increased in COVID-19
[22]. In addition, the upregulation of chemoattractants, such as CXCL-8, promotes the infiltration of neutrophils in the lung, thus contributing to ROS generation and protease activation, which inflict mitochondria damage
[23].
3.2. COVID-19: Disruption in the Mitochondrial Regulation of Iron Homeostasis
After being recollected, the iron is stored as ferritin in the mitochondria for multiple biochemical functions, such as mt-ROS formation
[24]. Ferritin is an intracellular protein, whose function is to regulate the iron deficiency or its overload via mitoferrin carriers. The disruption of cellular levels, mitochondrial iron metabolism, and mitoferrin carriers leads to hyperferritinemia, associated with hyper-inflammation and mt-DNA damage
[25], which exacerbates the cellular OS and the massive release of inflammatory mediators, ROS, and free radicals. In addition, the disrupting mitochondrial homeostasis caused by the hyperferritinemia drives mitochondrial respiration from an aerobic into an anaerobic state, which favors pyruvate reduction into lactate, creating an important amount of LDH
[26]. This metabolism alteration is associated with an increased conversion of NADH into NAD
+, which inhibits the mitochondrial metabolism in an out-of-control positive feedback that intensifies the inflammation response
[27].
3.3. Mitochondrial Disfunction in the Hypercoagulability State Associated with COVID-19 Severity
Coagulation abnormalities have been associated with the severity and aggravation of COVID-19 patients. Thrombocytopenia is reported to be more prominent in severe patients in comparison with non-severe cases
[28] and is used as a prognosis factor to identify patients at risk of developing severe conditions, such as disseminated intravascular coagulation (DIC). The relation of platelets and hypercoagulable states presented in COVID-19 is associated with the absence of the genomic DNA of the platelets, causing them to preserve organelles, such as mitochondria, to maintain their structure and function. The accumulation of five-to-eight mitochondria is critical for vital platelet functions, such as metabolism, aerobic respiration, ROS generation, reduction of mitochondrial membrane potential, and platelet apoptosis
[29]. Mitochondrial dysfunction in COVID-19 altered platelet survival and apoptosis, raising the mt-ROS production, which leads to severe OS, an altered mitochondrial membrane potential, and a reduced ATP production, thus increasing the risk of thrombus formation
[8].
These findings contribute important evidence suggesting that mitochondrial disruption interferes with effective immune responses, increases the inflammatory state, exacerbates OS, and predisposes patients to an increased COVID-19 severity. Nevertheless, the mechanism involved in other viral infections, such as the ER associated with an altered Ca2+ excretion and disfunction of MAMs, has not been fully addressed in the COVID-19 pathology.
3.4. Role of the Mitochondrial Dysfunction in Arterial Hypertension and Diabetes: Risk Factors Associated with COVID-19
Mitochondrial dynamic and function is affected by the changes, alterations or needs in response to metabolic signals. The metabolic changes in multiple diseases, such as hypertension and diabetes, affect the form, number, and function of the mitochondria, which consequently intervene in the organ’s functionality.
The hypertension-related cardiac hypertrophy has been associated with alteration of the ETC, metabolic substrate utilization changes and ATP synthesis
[30], identifying that proteins involved in mitochondrial OXPHOS were overexpressed whereas the α-subunit of the mitochondrial precursor of ATP synthesis was down-regulated
[31]. These results suggest that hypertrophic hearts are related with an altered mitochondrial energetic metabolism, including decreased ATP production and cellular respiration.
Moreover, the increased ROS generation caused by reduced antioxidant capacity in cardiovascular, renal, and nervous systems, has been identified as an important role in the vascular remodeling, vasoconstrictor, and vasodilator responses and in altered vascular mechanics associated with hypertension
[32]. Similarly, hyperglycemia and insulin resistance are also linked to excess ROS production
[33], that subsequently trigger both mitochondrial mediated cell death and the degradation of mt-DNA
[34].
Pancreas cell alteration and deficient insulin secretion play an important role in the development of insulin resistance and diabetes; further, alterations of mitochondrial dynamics have been related to disruption of pancreatic beta cells function
[35]. Opa1, a GTPase associated in the mitochondrial fusion machinery, is required for an adequate function of the respiratory chain in pancreatic beta cells. Thus, the ablation of Opa1 cause mitochondrial fragmentation and death of pancreatic beta cells
[36], associated with reduced insulin secretion and impaired systemic glucose homeostasis. Further, defects in glucose-stimulated mitochondrial ATP production in beta cells deficient in Opa1 may result from OXPHOS alterations, exacerbating the deficient insulin secretion.
The relation of mitochondrial functionality in the development of these diseases, associated with the severity of COVID-19, emphasizes the importance of the mitochondria in the exacerbation of this viral disease.
3.5. Abnormal Functioning of Mitochondria and Release of mt-DNA as DAMPs
During SARS-CoV-2 infection, hypoxia cause a switch to aerobic glycolytic respiration, which reduces the ETC Complex I–IV activity and alters the OXPHOS and mitochondrial function, inducing a condition of hyperglycemia, elevated ATP production, and increased oxygen consumption
[37]. These altered metabolic pathways lead to an increase in cytoplasmic and mitochondrial ROS. The failure of damaged and senescent mitochondria to clear the ROS may damage the mitochondrial structure, biogenesis units, and lipid membranes, and finally, it may generate altered mt-DNA levels
[38]. Furthermore, as the virus uses mitochondrial machinery to replicate, the integrity of the mitochondrial membrane is lost, releasing mt-DNA into the cytosol and circulation
[8]. These circulating mt-DNA fragments (DAMPs), possessing functions similar to the PAMPs released by the microbes, are identified by PRRs
[7]. The most well-known PRRs are TLRs, which are membrane-bound or intracellular receptors expressed by all innate immune cells with several different subtypes: TLR4 detects lipopolysaccharides (LPS), a Gram-negative bacteria membrane component; TLR2 detects cell wall components, such as peptidoglycan and lipopeptides from Gram-positive bacteria; and TLR9 detects unmethylated CpG DNA motifs of the mitochondrial genetic component
[39]. The TLR9 signaling pathway results in the activation of NF-kβ and mitogen-activated protein kinases (MAPKs), which trigger a pro-inflammatory response, initially with the activation of immune cells. Activated innate immunity induces gene transcription, which leads to the production of signal molecules, such as pro-inflammatory cytokines (IL-6 and IL-8), prostaglandins, interleukins, and chemokines, and it also activates the complement system, evoking the inflammatory cascade
[40].
Another function of the DAMPs is to activate the NF-Kβ pathway and inflammasomes, mainly the NLRP3 inflammasome
[41]. The NLRP3 inflammasome, containing the pyrin domain-containing protein 3 (NLRP3), is activated by a large range of mitochondrial ligands, such as mt-DNA, ATP, and mt-ROS, and by inducing mitochondrial dysfunction. After the activation of NLRP3, an adaptor protein apoptosis-associated speck-like protein recruits caspase-1 to the inflammasome through a caspase recruitment domain. When caspase-1 is activated, it cleaves pro-interleukin 1B and pro-interleukin 18 to their biological forms
[42], leading to an inflammation cascade. Mt-DNA, as a DAMP, could also affect immune responses by the cyclic AMP-GMP synthase (cGAS) pathway. The cGAS pathway, as part of the innate immune system, detects the presence of mt-DNA in the cytosol, triggering type 1 IFN inflammatory responses
[43]. cGAS, its second messenger cyclic GMP-AMP (cGAMP) and the sensor of cGAMP, whose main function is the stimulation of interferon genes (STING), are a major sensing pathway for cytosolic DNA
[44]. The stimulation of this pathway induces NF-kB activation, controlling the transcription of pro-inflammatory cytokines and chemokines
[45].
Mitochondrial DAMPs are an important element in the infection detection, stimulation, and maintenance of immune response against pathogens
[46]. However, the sensing of mitochondrial DAMPs may also cause an undesirable immune response and result in an unwanted inflammation. In conditions of massive cell damage, such as COVID-19 disease, protective mitochondrial mechanisms, as well as mitophagy processes in which damaged mitochondria are phagocyted, may not work or may be overwhelmed, inducing the release of mitochondrial DAMPs into the cytosol and circulation in overwhelming amounts. These actions may trigger an unwanted pro-inflammatory response
[47]. These mechanisms exacerbate tissue dysfunction by involving metabolic, endocrine, cardiovascular, coagulation, immune, and thermoregulatory pathways, and they are also responsible for systemic effects and multi-organ failure
[48].
From the data presented in the study of Indraneel Mittra et al., the authors refer to the possibility that exogenous nucleic acid (ENAs), such as mt-DNA, can be taken up by tissue cells. This proposal is similar to the hypothesis of “geno-methastasis”, which claims that once inside cells, ENAs have several biological actions, such as inducing genetic, cellular, and chromosomal damage. ENAs are certainly capable of activating both innate and adaptive immune systems and inducing a sterile inflammatory response
[49][50]. These findings highlight the importance of the increase of circulating free mt-DNA fragments and the decrease of intact mt-DNA as a consequence of the SARS-CoV-2 infection.
 +1 point
+1 point