1000/1000
Hot
Most Recent
 +1 point
+1 point
Hepatocellular carcinoma (HCC) is a difficult to treat liver cancer that generally arises in individuals suffering from alcoholic or non-alcoholic fatty liver diseases. Inflammation, tissue injury and fibrosis are important precursors of HCC. Translocation of microbial- and danger-associated molecular patterns (MAMPs and DAMPs) from the gut to the liver elicits profound chronic inflammation, leading to severe hepatic injury and eventually HCC progression.
The liver is intimately linked to the gut and represents a critical metabolic hub involved in digestion, detoxification and clearance of microbial products. Its building blocks are the hepatic lobules organized around central veins and portal triads, consisting of a portal vein, a hepatic artery and a bile duct. The portal vein delivers 80% of the total liver blood supply, while the remaining 20%, i.e., the oxygenated blood, flows through the hepatic artery. Upon mixing, the blood flows across the lobule through the hepatic sinusoids and drains into the central veins, while the bile flows in the opposite direction via the bile canaliculi. Such an organization establishes gradients of oxygen and metabolites and creates a liver zonation. The hepatocytes, endothelial and immune cells of the liver are aligned along this vasculature, and their spatial distribution along the liver zonation dictates their phenotypes and functions.
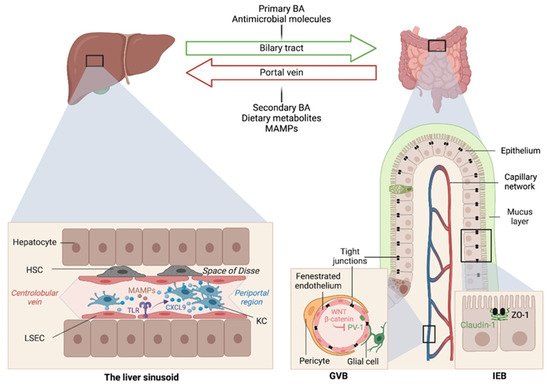
At birth, we are colonized by a collection of microorganisms, including bacteria, fungi, viruses and archaea, that outnumber our human cells by 10:1 and provide 100-fold as many genes as found in our human genome [1]. The gut microbiota provide essential functions for our digestion and nutrient absorption, including the breakdown of indigestible carbohydrates, the synthesis of vitamins and the deconjugation of primary BA, and in shaping our mucosal immune system.
Altered intestinal microbiota, accompanied by small intestinal bacterial overgrowth (SIBO), are observed in liver chronic inflammatory diseases [2][3][4], cirrhosis [5][6][7][8][9] and HCC [10], and gut microbiome metagenomic signatures have been identified in patients with NAFLD [11], cirrhosis [3] and HCC [12]. For instance, Boursier et al. reported that fecal Bacteroides and Ruminococcus were independently associated with NASH and fibrosis (stage 2 or higher), respectively, while Prevotella was depleted in these conditions [7]. Loomba and colleagues showed an increased abundance of Bacteroides vulgatus and Escherichia coli in NAFLD patients with advanced fibrosis [11]. Of note, E. coli is the predominant bacterium detected by culture in NAFLD patients exhibiting SIBO [2]. In mice, prolonged high dietary cholesterol feeding caused spontaneous NAFLD and progression to HCC development, which was associated with gut-microbiota dysbiosis [13]. Importantly, treatment with a cholesterol-lowering drug restored the microbial ecology and prevented disease development in mice [13]. In mouse models of NASH or obesity-induced HCC, Akkermansia spp., Prevotella spp. and Lactobacillus spp. are decreased, while Bacteroides spp., Clostridium spp. and Ruminococcus spp. are elevated [14][15][16] (Figure 2). Last, besides bacteria, the intestinal microbiota diversity is reduced in patients with liver diseases, particularly ALD patients, in whom candida dominates [17]
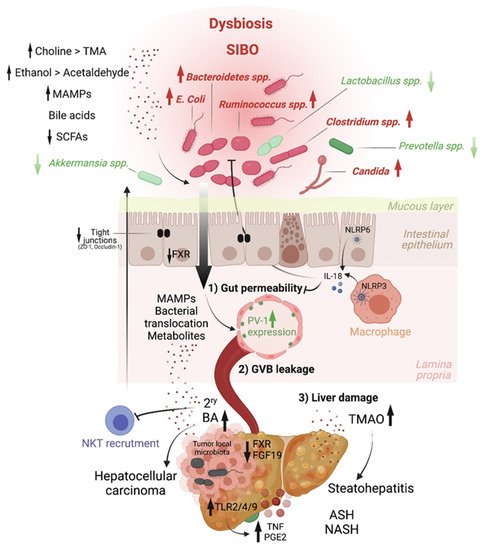
Early studies have reported a link between liver diseases and disrupted intestinal epithelial cell (IEC) barrier integrity. For instance, NAFLD patients with no significant alcohol consumption were shown to exhibit disrupted IEC tight junctions and increased prevalence of SIBO, which correlated with the severity of steatosis [21]. Similarly, both in vitro and in vivo studies demonstrated the impact of alcohol on enhancing intestinal permeability by altering the expression of the tight junction proteins zonula occludens-1 (ZO-1) and claudin-1 [22][23]. Consistently, occludin deficiency in mice led to a more severe ALD phenotype [23]. Besides the IEC barrier, a gut vascular barrier (GVB), as described by the Rescigno lab [24], also restricts liver injury and the translocation of bacteria or bacterial products into the systemic circulation. The GVB is composed of intestinal endothelial cells closely associated with pericytes and enteric glial cells and is thought to provide an additional protective layer that shields the liver from microbial moieties and inflammatory damage [25]. The integrity of the GVB is sustained by the activation of the WNT/β-catenin pathway, which inhibits the expression of the plasmalemma-vesicle-associated protein 1 (PV-1) [24], a membrane glycoprotein associated with the structure of fenestrated endothelia [26], and upregulated in leaky GVB [24] (Figure 1).
It is posited that gut microbiota dysbiosis results in reduced BA-mediated stimulation of the farnesoid X receptor (FXR) that drives β-catenin activation in endothelial cells. This has been demonstrated in mouse models of NASH induced by a HFD or a choline and methionine deficient (MCD) diet [27], and a mouse model of cirrhosis induced by bile duct ligation combined with carbon tetrachloride (CCL4)-mediated liver injury [28]. In both models, intestinal permeability led to microbial translocation from the gut to the liver, and FXR agonists, such as the BA analog obeticholic acid (OCA), conferred protection against GVB disruption, limited bacterial translocation and ameliorated liver pathology[29][30].
Short Chain Fatty Acids. SCFAs, which are produced by bacterial fermentation of dietary fibers, have been recently implicated in HCC. SCFAs, including butyrate, propionate and acetate, are generally associated with metabolic health [52]. For instance, in a randomized controlled trial that administered the fermentable fiber inulin coupled to a propionate ester to overweight adults reported decreased abdominal adiposity, lipid accumulation in the liver and body weight gain compared to controls receiving inulin alone [53].
This effect could be mediated by enhanced fat acid oxidation and energy metabolism, as shown in a second trial in which a mixture of SCFAs was infused in the colon of overweight or obese men [54]. Butyrate and propionate production are also altered in ALD. The reduction of butyrate is associated with a weakness of intestinal permeability, while the administration of butyrate in the form of tributyrin reduced intestinal permeability and subsequent liver injury in ethanol-fed mice [55]. However, this notion has been recently challenged by the group of Vijay-Kumar, who showed that large amounts of SCFA, particularly butyrate in a context of dysbiosis, may instead create a tumor-promoting environment. They reported that, while inulin protected mice from obesity, a longer period of inulin feeding, i.e., over 6 months, promoted cholestatic HCC in 40% of TLR5-deficient mice with a pre-existing dysbiotic microbiota [56]. Soluble-fiber-induced HCC was shown to be microbiota-dependent and transmissible to wild-type (WT) mice in cohousing or cross-fostering experiments. More importantly, interventions that deplete butyrate-producing bacteria, e.g., with metronidazole, inhibit gut fermentation, e.g., via supplementation of plant-derived -acids, exclude soluble fiber from the diet, or prevent enterohepatic recycling of BAs with cholestyramine reverted inulin-induced HCC in these mice [56].
Choline. Another host–microbiota-controlled metabolite is the macronutrient choline. Choline processing into phosphatidylcholine by the host prevents steatosis. Indeed, feeding mice a choline-deficient diet is a classical model of NASH. Alternatively, choline can be converted into trimethylamine (TMA) by intestinal bacteria, and it is then further metabolized in the liver into trimethylamine-N-oxide (TMAO). In contrast to phosphatidylcholine, TMAO is at the origin of hepatic steatosis by promoting triglyceride accumulation (Figure 2). Increased systemic circulation of TMAO is associated with a reduced level of phosphatidylcholine, and this imbalance is characteristic of patients with NAFLD [57] and NASH [58][59].
Ethanol. Ethanol metabolism greatly impacts HCC development through different mechanisms. The commensal microorganisms express genes that can ferment dietary sugars into ethanol that is absorbed by the gastrointestinal tract by simple diffusion. Moreover, liver cells, enterocytes and gut microbiota components express alcohol dehydrogenase, which co-metabolizes ethanol into toxic acetaldehyde that is then converted by CYP2E1—if this pathway is not saturated—to acetate. Acetaldehyde has been involved in weakening tight junctions of the intestinal epithelial barrier [23], eliciting gut barrier permeability and enabling translocation of microbial products, as shown in ethanol-fed mice [60]. In addition, ethanol exacerbates oxidative stress and hepatic inflammation and is a known carcinogen [61].
While much progress has been made in this field, for instance, in cataloguing enriched or depleted microorganisms in health versus disease, we now need to move to next-generation studies of the functional microbiome in order to be able to design microbial strategies to counter liver diseases and HCC.





