Although being usually characterized by an indolent course, a variable portion of patients manifests symptoms, mainly vision loss and other ophthalmological symptoms, but also precocious puberty or neurological manifestations
. The comprehension of the biology of these tumors has improved significantly over the last few years but challenges still remain: (i) the risk assessment in asymptomatic patients remains demanding, owing to the lack of valid biomarkers and the absence of prospective studies that may help in prognosis definition; (ii) the young age of this unique at-risk population and the learning disabilities that frequently coexist complicate the development of an effective OPG screening; (iii) treatments able to prevent or recover vision loss in patients with OPG are still not available.
2.1. Prevalence, Clinical Features and Natural History of OPG
OPG is the most common central nervous system neoplasia detected in pediatric patients affected by NF1, with an estimated prevalence ranging from 15% to 20%
[6][9][10][11][12][13][14][15][16][17]. In the majority of cases, NF1-associated OPGs are classified as WHO grade I pilocytic astrocytomas and only 30–50% of patients show signs or symptoms correlated with the tumor
[11][16][17][18][19][20]; in addition, they usually present at a younger age compared to sporadic OPGs in the general population
[21] and are characterized by an indolent course, with only one-third of the affected patients requiring a specific treatment
[20]. Some studies reported a higher prevalence of OPGs among females
[6][10][15][22][23], but several others did not observe such a difference according to sex
[16][17][24][25].
A recent study evaluated the prevalence of OPG in an unselected cohort of patients with NF1 followed up in a single NF clinic in Germany between 2003 and 2015; all patients were offered whole-body and head MRIs regardless of the presence of symptoms suggestive of OPG
[17]. The authors identified a particularly high prevalence of asymptomatic OPG among children younger than 10 years (approximately 20%), which dropped to 5–10% in the group of patients aged 10–19.9 years; on the other hand, the prevalence of symptomatic OPG was lower than 5% in patients younger than 10 and approximately 5% in those aged 10-19.9 years
[17]. The prevalence of asymptomatic OPG in children under 10 years was higher than in other studies, but this may be due to the use of different radiologic criteria for the diagnosis; for example, a T2 hyperintensity of the optic nerve was classified as OPG by Sellmer et al.
[17] without taking into consideration its thickness or tortuosity.
Our experience at the NF1 clinic of the University Hospital of Padova (Italy) was also published
[6]. We analyzed a cohort of 414 consecutive patients affected by NF1 who were first evaluated before the age of 6 years and without a previous diagnosis of OPG; the inclusion criteria were chosen to avoid bias in patients’ selection and the mean duration of follow-up was 11.9 years. In our clinic, screening MRI is not systematically performed in all patients. A total of 52 patients (12.6%) developed OPG during their follow-up, with an estimated cumulative incidence of 15.4% at the age of 15 (Kaplan–Meier analysis). In particular, 25 children were diagnosed with OPG after brain and orbit MRI was performed because of the presence of signs and/or symptoms suggestive of OPG; the remaining 26 did not show symptoms related to OPG at the diagnosis and underwent brain imaging because of other clinical indications (including macrocephaly, developmental delay, headache, seizures, plexiform/orbital neurofibromas of the face/neck, brain ischemia) or for screening surveillance in other hospitals.
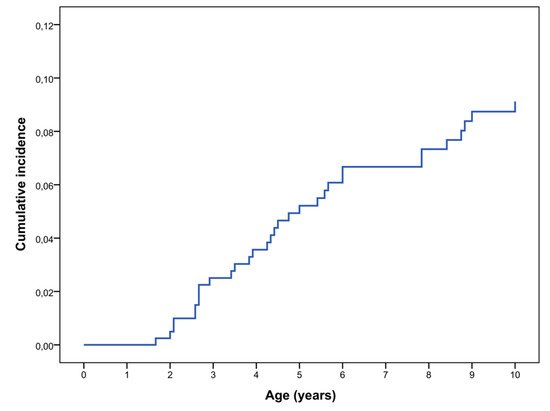
We have re-evaluated these data and calculated the cumulative incidence of symptomatic OPG in the first decade of life; OPG was defined symptomatic if associated with any of the following anomalies: decreased visual acuity, ophthalmoscopic anomalies, optical coherence tomography (OCT) alterations, proptosis, precocious puberty, or other signs/symptom related to the tumor. In our cohort the rate of symptomatic OPG was 9.1% at 10 years of age (Kaplan–Meier analysis) (Figure 1). We excluded all the patients who underwent brain MRI scans for other clinical indications and were found to have OPG; anyway, none of these patients required any treatment during their follow up.
Figure 1. Cumulative incidence of symptomatic OPG in NF1 diagnosed patients in the first decade of life (N = 414) (Kaplan–Meier analysis).
The prevalence of symptomatic OPG is comparable to that reported in other previous studies. However, we could not calculate the prevalence of asymptomatic OPG in our cohort since brain MRI was not systematically performed in all patients for screening purpose.
In the literature, the mean age at diagnosis ranges from 3 to 6 years
[6][10][11][12][16], with estimates in the lower part of this range in studies with a systematic MRI screening program for NF1 patients
[15][16][22][26]. OPGs rarely grow after 10 years of age
[27] and few cases of adolescent or adult patients with a late onset or late-progressive OPG have been reported in the literature
[25][28]. Additionally, OPG prevalence among adult patients was evaluated in few studies and was estimated to be approximately 5%
[17][29].
Asymptomatic OPGs are prevalent in children with NF1 who are younger than 10 years and their frequency decreases with increasing age until adulthood
[17]; conversely, the prevalence of symptomatic tumors (3% to 5% of patients with NF1) remains stable
[16][17]. There are several hypotheses to explain the reduction of the prevalence of asymptomatic OPGs among adult patients: first of all, tumors may undergo spontaneous regression, as documented by several clinical reports
[17][30][31][32]; additionally, since the histopathological examination of OPGs is rarely performed, there is the possibility that some of them are not true neoplasms.
Symptomatic OPG may present with decreased visual acuity, visual field deficits, optic disc swelling and/or disc pallor, proptosis, visual evoked potentials (VEPs) alterations, optical coherence tomography (OCT) abnormalities, strabismus, nystagmus, precious puberty, and various neurologic signs
[6][20][33]. Signs and symptoms depend on the location of OPGs, which can arise anywhere along the optic pathway: intraorbital optic nerves, prechiasmatic optic nerves, chiasm, optic tracts and optic radiations
[34]. Approximately 45 to 65% of OPGs involve only the prechiasmatic pathway, while 25% to 50% involve the chiasmatic region with or without the optic nerves; the optic tracts and radiations are affected (with or without the chiasmatic and/or prechiasmatic regions) in the remaining cases
[6][15][16][17][25]. Symptomatic OPGs are associated with significant morbidity but there is no evidence that they are risk factors for premature death in NF1 patients
[35].
Some prognostic factors associated with an increased risk of OPG clinical progression have been proposed. Females have been shown to lose vision and to require treatment in a higher proportion than males
[15][24][36]; moreover, gliomas diagnosed in children younger than 2 years and older than 8–10 years are more aggressive than those presenting between 2–8 years of age
[28][29][37][38]; finally, retrochiasmatic OPGs seems to have a more aggressive clinical behavior compared to prechiasmatic and chiasmatic ones
[19][29][37]. However, conflicting results have been reported in the literature and currently no clear prognostic factors are available, making the clinical evolution of OPG unpredictable in the majority of cases
[6][39].
2.2. Genotype-phenotype Correlations in OPG
The mutational spectrum at the
NF1 locus has been examined by a number of studies in order to evaluate the risk of OPG in NF1 asymptomatic patients. Despite the small number of recurrent mutations and the well-known intrafamilial variability in the expressivity of the disease, advances in molecular technologies in the last 20 years, with an increasing number of genotyped patients, have allowed to establish a growing number of genotype-phenotype correlations, with relevant consequences for the clinical management
[40][41][42][43][44][45][46]. However, the variable timing, nature and location of
NF1 somatic mutations on the other allele significantly contribute to disease severity, making identification of such correlations challenging.
The relationship between OPG development and
NF1 genotype is still under debate. Some pathogenic variants in
NF1, including the p.Met992del deletion detected in 170 individuals and the substitutions at codon 1809 identified in 98 patients, have never been reported in association with symptomatic OPG
[41][43]; conversely, these tumors were found to be more prevalent in patients harboring missense variants affecting codons 844–848
[44]. Previous works focused on the risk of OPG in NF1 patients and reported conflicting results on the association between tumor development and mutation position, with some studies reporting an increased risk of OPG in patients harboring mutations in the 5′tertile of
NF1 (exons 1–21)
[47][48], while others not confirming this finding
[49]. In order to bypass limitations attributed to the small sample size, further adequately powered analyses on combined studies finally confirmed the correlation between the presence of 5′-end gene mutations and the likely of developing OPG (OR 2%, CI: 1.22–3.29;
p = 0.006)
[50][51][52]. Moreover, Xu and colleagues
[51] elegantly showed that mutations clustering in specific regions of neurofibromin confer different risks of OPG: pathogenic variants correlate with a higher or lower risk of developing these tumors when located in the cysteine/serine-rich (CSRD, amino acids 543–909) or HEAT-like repeat region (HLR, amino acids 1825–2428) domains, respectively, despite the specific mutation type. These findings indicate that the
NF1 germline mutation might become a useful biomarker to predict OPG development and emphasize the importance of a better comprehension of the functions of neurofibromin. However, sensitivity and specificity of these correlations are currently not sufficient to provide an actual utility in the clinical setting
[52], and further studies, evaluating other risk factors beyond the germline mutation, are warranted.
 +1 point
+1 point