1. Introduction
Failed drug therapy due to unintended drug-drug interactions occurs widely in clinical medicine and could impact drug efficacy and safety [1][2][3]. According to the World Health Organization, the annual financial cost of medically-related harm globally is approximately $42 billion USD [4]. In order to exert a therapeutic effect, drugs must be absorbed via a certain route of administration, such as oral, intravenous, intramuscular, nasal and subcutaneous, among which oral delivery is the most preferred method due to several well-recognized reasons, including convenience, non-invasiveness, extended drug release, suitability for long-acting medication and a long shelf-life [5][6], but the liver and small intestine constitute the main sites of drug metabolism, leading to pharmacokinetic (PK) variability (i.e., different drug concentrations in the blood or at sites of action) [7][8]. Special populations, such as the elderly, children, women, pregnancy, hospitalized patients and even certain ethnicities, are particularly vulnerable to certain prescribed oral medication(s), because their physiological differences (e.g., metabolism) and/or ongoing life circumstances (e.g., polypharmacy, comorbidities) may result in unpredictable drug-drug interactions [9].
Before reaching the target site of action, orally administered drugs encounter a host of obstacles in the gastrointestinal tract (GIT), such as a substantially changing pH in the stomach, upper and lower intestinal segments, extensive enzymatic degradation (e.g., lipase, trypsin, amylase), varied GI motility (e.g., gastric emptying, peristalsis), complex bacterial diversity and physical barriers of the mucus and mucosal layers [10][11]. Among those complex GIT environmental factors, pre-systemic metabolism of Cytochrome P450 (CYPs) enzymes expressed in the intestine and liver is the main factor that significantly contributes to the variability in drug response [12]. Particularly, out of all the CYPs involved in human drug biotransformation, CYP3A4 is the most important oxidation enzyme by virtue of the fact that at least 50% of marketed drugs metabolized by CYPs are metabolized by CYP3A4 [8][13]. The amount and activity of CYP3A4 is considerably regulated by inflammation, fasting state and a broad spectrum of xenobiotics, including top prescribed pharmaceuticals (e.g., midazolam, felodipine), common foods (e.g., grapefruit juice) and widely used herbal medicines (e.g., St. John wort (SJW)) [14][15][16] ( Figure 1 ). However, drug development selects the most effective and safe dose in the study population (not the individual) without any regard for such drug response variability due to CYP3A4 metabolism, as it is impractical to investigate many different doses in different patients. Yet, from a clinical medicine point of view, “one-dose-fits-all” regimen can be potentially dangerous for patients as huge inter- and intra-individual variability in CYP3A4 may lead to varied systemic drug concentrations, in turn, causing unpredictable therapeutic outcomes and intolerable adverse effects [17].
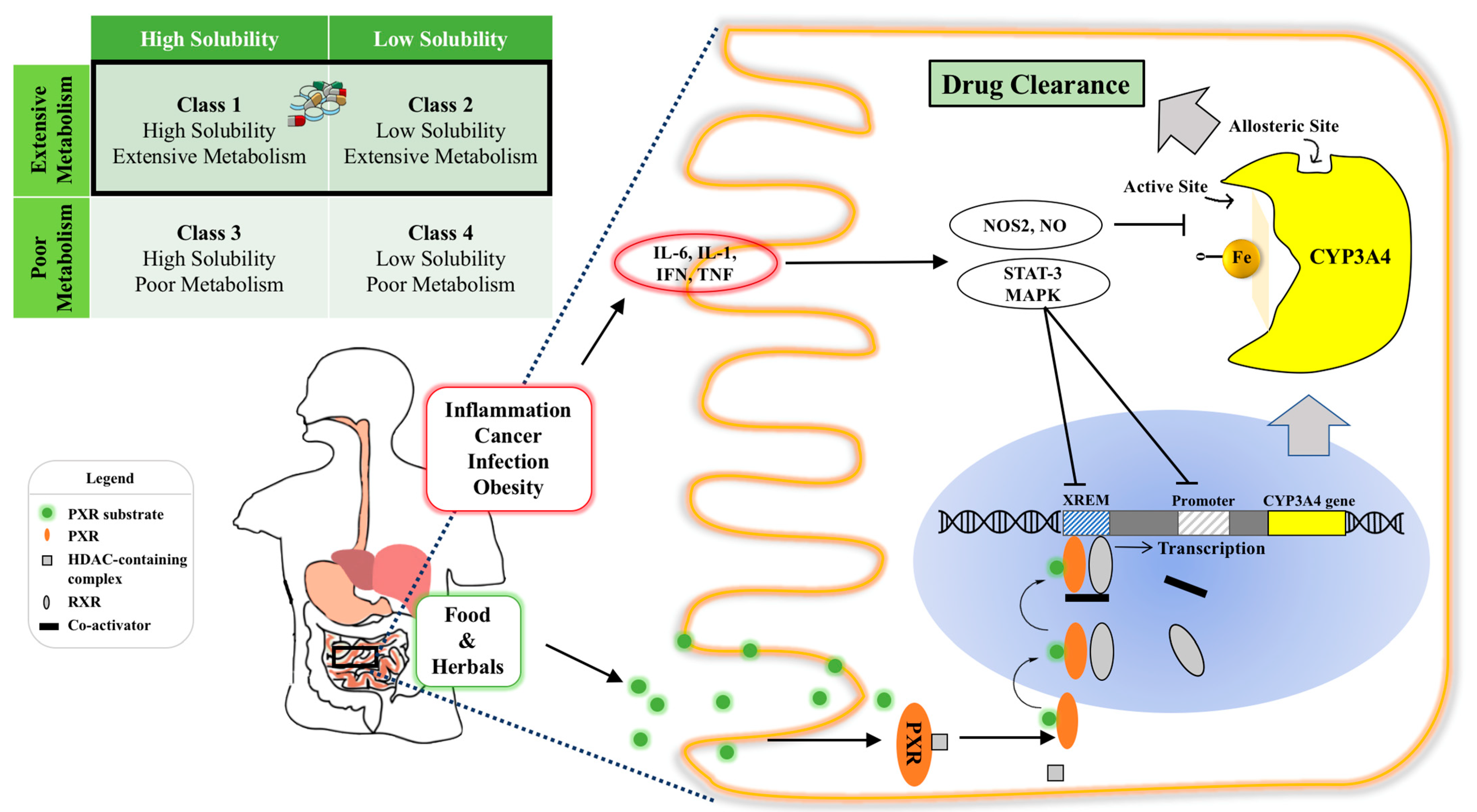
Figure 1. Illustration of intestinal CYP3A4 regulation and the effect of its content and activity variation on orally administered drugs. Top left: CYP3A4 ligands belong to BDDCS Class 1 and Class 2 with varied solubilities and extensive metabolism, highlighted by the black rectangle; Bottom panel: intra-enterocytic CYP3A4 regulation by endogenous factors under disease conditions and xenobiotics from oral intake (e.g., drugs or food constituents). Normally, a PXR ligand enters the enterocyte and binds to PXR intracellularly. This then dimerizes with retinoid X receptor (RXR) and binds to the xenobiotic response enhancer module (XREM) to upregulate the CYP3A4 gene. Systemic inflammatory conditions such as cancer, infection and obesity increase circulating cytokines, such as IL-6, which activate the STAT3-MAPK pathway to downregulate CYP3A4 gene regulation. Intra- and inter-individual CYP3A4 variation can cause varied drug clearance, resulting in undesirable toxicity and ineffective therapy of drugs with a narrow therapeutic index. Abbreviation: RXR, retinoid X receptor; XREM, xenobiotic response enhancer module (XREM); STAT: signal transducer and activator of transcription-3; MAPK: a family of signaling cascade including Jun N-terminal kinase and mitogen-activated protein kinase 1 pathways.
Despite incongruity between industrial and clinical sectors in how to determine drug dosing, for most drugs, the prediction of their in vivo efficacy and toxicity relies on free (i.e., bioavailable) drug concentration at the site of action [18]. However, before reaching systemic circulation, the passage of orally administered drugs through the GIT could be attenuated by the presence of drug transporters (e.g., P-glycoprotein (P-gp)) and metabolizing enzymes (e.g., CYP3A4) in the small intestine. The additional barrier of first-pass metabolism, as the portal vein flows through the liver, also contributes to drug elimination.
Once systemically available, drugs may partition into other tissues such as the lung and heart, depending on their physiochemical properties such as lipophilicity. In addition, food intake has a strong effect on the extent of oral drug absorption
[19][20][21]. Particularly, postprandial state with a high fat meal can have both positive and negative impact on the drug bioavailability by altering the dissolution, the rate and extent of absorption
[22]. Ingested dietary lipids (e.g., triglyceride, monoglyceride, fatty acids) are known to increase dissolution and solubilization of lipophilic compounds with log P > 5, modulate GIT transit and inhibit CYPs activity
[22][23]. Because of this, the U.S. Food and Drug Administration (FDA) has issued guidance for “Food-Effect Bioavailability and Fed Bioequivalence Studies” to label some medications that are strongly influenced by a meal
[24]. All of these factors could confound the predication of a therapeutically relevant dose, and partially attribute to the decline in accumulative clinical success rate (i.e., ~11.6%)
[25][26]. As a result, the question of whether adequate drug concentrations at the target have been achieved may underpin variability in drug response.
The influence of those physiological GIT barriers (e.g., CYP3A4, pH) as well as extrinsic regulatory variables (e.g., diet, health conditions) on absorbed drug concentrations can be largely determined by the properties of the oral dosage form, which in turn depends on its design and manufacture
[27]. For example, in a randomized crossover study, Mueller et al., compared two marketed cyclosporin formulations, Sandimmune vs. Sandimmune Neoral
®, and found that the long chain triglyceride-based Neoral
® was much less influenced by fat-rich meals, indicating that lipid-based formulations to deliver the drug was advantageous when individualizing a dosage regimen
[28][29]. Especially, lipid-based formulations, which account for approximately 2–4% of the pharmaceutical market, represent one of the most popular approaches to improve the solubilization of poorly water-soluble compounds and to overcome the GIT barriers. Pharmaceutical grade lipid excipients in lipid-based formulations are physiological or physiologically related, including fatty acids (e.g., oleic acid, myristic acid), ethyl esters (e.g., ethyl oleate), triglycerides of long- or medium- chain fatty acids (e.g., corn oil, Miglyol
®), and non-digestible mineral oil
[30]. The intraluminal process (e.g., bile salts) of ingested lipids from formulations forms solubilised phases to facilitate the drug absorption
[30][31].
Among diverse lipid-based formulations, lipid-based nanosystems (LNS) have been an ever-growing segment of pharmaceutics that primarily involves FDA’s Biopharmaceutics Classification System (BCS) Class 2 & 4 drugs with low solubility and/or poor permeability
[32]. LNS encompasses various subtypes with different nanostructures, including solid lipid nanoparticles (SLN), liposomes, nanostructured lipid carriers (NLC), polymer-lipid hybrid nanoparticles (PLN), and self-nanoemulsifying drug delivery systems (SNEDDS)
[33][34][35][36][37]. Because of combined attributes of both lipidic carriers and nanosized particles, LNS can facilitate the delivery of active pharmaceutical ingredients (API) into the blood circulation via either the hepatic portal vein or the GIT lymphatic system
[38]. Particularly, for LNS containing triglycerides, cholesterol esters or long chain fatty acids, they are incorporated into the large lipoprotein chylomicrons (75–1200 nm) upon re-esterification and preferentially trafficked via the intestinal lymphatic system, thus effectively evading first-pass metabolism in the liver
[39]. In recent years, an increasing body of evidence has shown that certain components (e.g., surfactants, lipids and polymers) used in LNS can reduce intra-enterocyte metabolism
[40]. Using CYP3A4 substrate midazolam as the probe, studies of effects of common surfactants, co-solvents and oils (e.g., Tween, PEG, poloxamer, oleic acid) on the drug disposition found 68.2% of tested excipients significantly inhibited CYP3A4 biotransformation activities
[41][42]. In addition, with advancing nanotechnology, LNS can be adapted by modulating their lipid composition and decorating them with polymers or ligands to target different regions of the GIT, in order to achieve a controlled, sustained, and targeted drug delivery
[43].
2. LNS Strategies of Overcoming Pre-Systemic CYP3A4 Metabolism
Many drugs are victims of CYP3A4 interactions. Unfortunately, interactions with CYP3A4 restrict the clinical use for these drugs that ultimately impact patient treatments. If these drugs can be formulated to avoid CYP3A4 binding, then they can be successfully used in therapy. Many lipid-based, polymeric and inorganic drug carriers have been used clinically and experimentally with great success to overcome the limitation of free therapeutics (e.g., solubility) and heterogenous biological barriers (e.g., systemically, microenvironmentally, and cellularly) across patient populations and diseases because of their capability of being engineered in a more personalized manner
[44].
Figure 2 illustrates three potential approaches of orally administered LNS to address enterocytic CYP3A4 metabolism and in turn, drug-response variability.
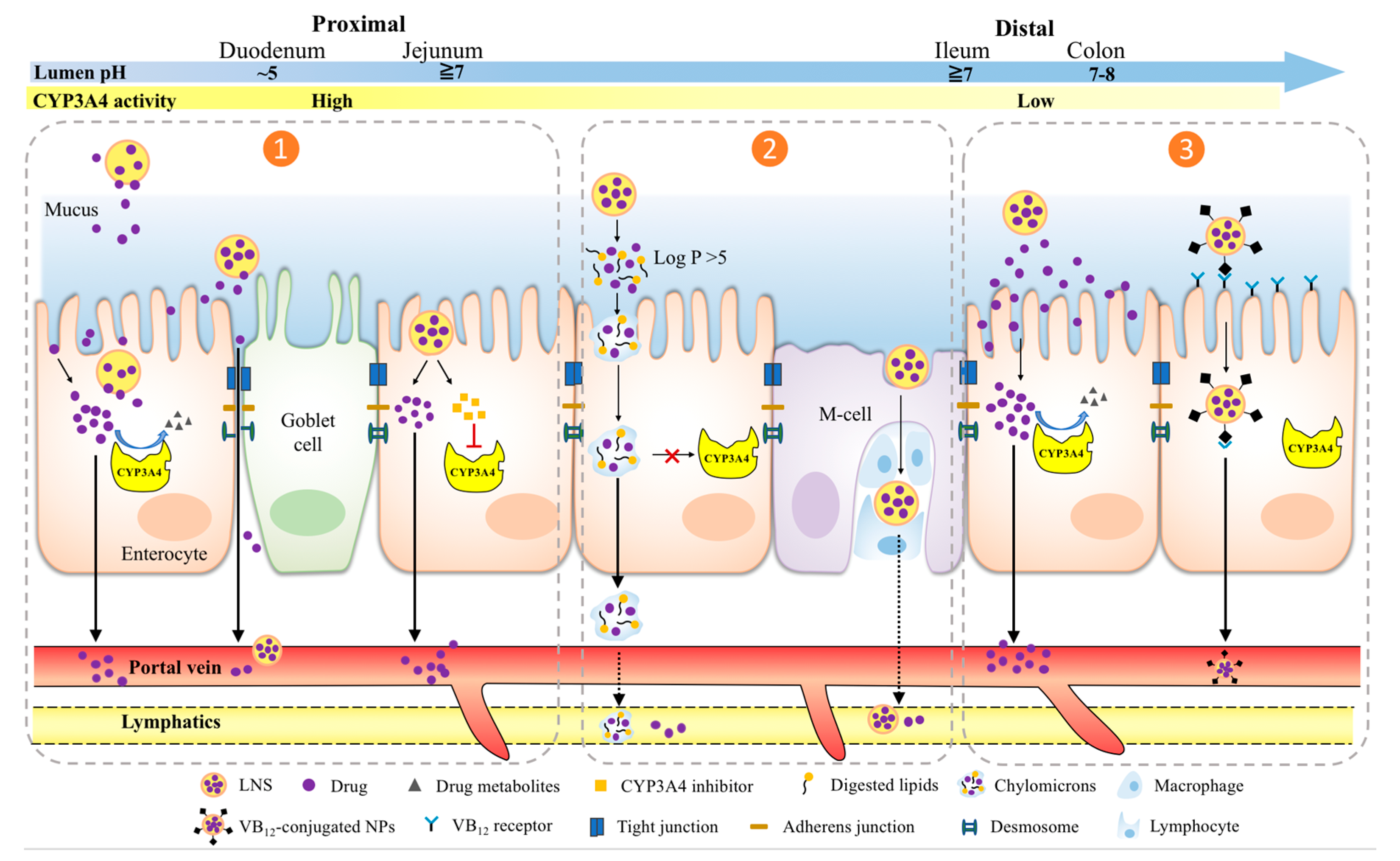
Figure 2. Different absorption strategies of LNS to address drug-response variability by intestinal CYP3A4 metabolism. Left panel (1): at the proximal small intestine, intra-enterocytic CYP3A4 activity can be locally saturated or inhibited by LNS delivered API or excipients; Middle panel (2): CYP3A4 metabolism is minimized via lymphatic drug transport; Right panel (3): Targeting the distal end of the small intestine where CYP3A4 activity is the least. Depending on the approach, various LNS types can be designed, from left to right: Mucoadhesive, CYP3A4 inhibitor-conjugated (combined), Chylomicron-mediated, M-cell mediated, pH-responsive and vitamin B12-mediated.
In
Table 1, select LNS examples and their delivered drugs based on BDDCS class are presented to demonstrate their mechanistic strategies of improving oral bioavailability
[45][46][47][48][49][50][51][52][53][54][55][56][57][58][59][60][61][62][63][64][65]. By taking advantage of existing physiological characteristics of the small intestines, the following six strategies have the potential to use nanoparticle (NPs) formulations to address intestinal CYP3A4 metabolism and to enhance the absorption of drugs and NPs across enterocytes: (1) incorporating mucoadhesive polymers or lipids that are attracted to the unstirred water layer adjacent to the intestinal epithelia, allowing drugs to be in close proximity which increases the flux into epithelial cells to overwhelm CYP3A4 metabolism; (2) CYP3A4 inhibitor-containing LNS locally inhibits intestinal CYP3A4 during transcytosis; (3) using highly lipophilic lipid NPs to traverse enterocytes straight into lymphatic vessels; (4) formulating LNS targeting M cell integrins for endocytosis that carries the drug to lymphatic vessels; (5) pH-sensitive formulation that selectively releases drug in the ileum where there is a lower expression of intestinal CYP3A4; (6) vitamin B12 targeting cubilin in the terminal ileum for absorption via receptor mediated endocytosis. Those strategies are GIT region dependent, so we discuss them separately below.
Table 1. Examples of orally administered LNS formulations for improving drug bioavailability.
| LNS a |
Delivery Mechanism |
Nanoformulations b |
Drug Payload |
BDDCS Class c |
Study Models |
Main Effects b |
Reference |
| Lipid NPs |
Mucoadhesive |
SLN |
Cyclosporin A |
Class 2 |
Young pig |
- -
-
low variation in drug bioavailability
|
[45] |
| Mucoadhesive |
VP16-NLC |
Etoposide |
Class 3 |
Rat intestinal membrane, Healthy rat |
- -
-
↑ intestine permeability
- -
-
↑ bioavailability by 1.8-fold compared to VP16 suspension
|
[46] |
| Clathrin-mediated endocytosis |
DRD-SLN |
Dronedarone hydrochloride |
Class 2 |
Healthy rat |
- -
-
↑ bioavailability by 2.68-fold compared to DRD suspension
- -
-
possible transport via lymphatic absorption
|
[47] |
| Lymphatic transport via chylomicrons |
EFV-SLN |
Efavirenz |
Class 2 |
Chylomicron blocking rat model, Mesenteric lymph duct cannulated rat model |
- -
-
↑ drug amount in the liver
- -
-
↑ accumulation in the spleen
|
[48] |
| Lymphatic transport via chylomicrons |
AT-NLC |
Atorvastatin |
Class 2 |
Both High-fat diet treated and health rats |
- -
-
↑ bioavailability by 3.6- and 2.1-fold compared to AT suspension and Lipitor®
- -
-
improved efficacy of AT by reducing serum levels of TC, TG and LDL
|
[49] |
| Portal vein and lymphatic pathway transport |
Darunavir-SLN |
Darunavir |
Class 2 |
Everted rat intestine, Chylomicron blocking rat model, Healthy rats |
- -
-
↑ bioavailability by 2-fold compared to marketed Darunavir tablet
- -
-
uptake by enterocyte via endocytosis
|
[50] |
| Portal vein and lymphatic pathway transport |
AM-SLNs |
Asenapine maleate |
Class 1 |
Caco-2 monolayer, Chylomicron blocking rat model |
- -
-
↑ bioavailability by 50.19-fold compared to AM dispersion
|
[51] |
| Lymphatic absorption |
CLA-SLN |
Clarithromycin |
Class 3 |
Healthy rat |
- -
-
↑ bioavailability by 5-fold compared to CLA suspension
|
[52] |
| Lymphatic absorption |
GEN-loaded SLN |
Genistein |
CYP3A4 inhibitor |
In vitro characterization of chylomicrons Caco-2 cells, Ex vivo porcine duodenum |
- -
-
2-fold increase in uptake of intestinal mucosa and enterocyte
|
[53] |
| Lymphatic transport via chylomicrons |
micelles |
5-demethylnobiletin |
N/A |
Caco-2 monolayer |
- -
-
↑ enterocytic metabolism of 5DN
- -
-
fatty acid types affected differently on intestinal uptake of the drug and chylomicron formation
|
[54] |
| Portal vein and lymphatic pathway transport |
CCN |
Candesartan cilexetil |
Class 4 |
Caco-2 monolayer, in situ single-pass intestine perfusion, ligated intestinal loop model, Healthy rats |
- -
-
internalized into enterocytes by clathrin-mediated endocytosis
- -
-
↑ permeability in the duodenum, jejunum and ileum
- -
-
↑ plasma AUC by 10-fold than free CC suspension
|
[55] |
| PLN |
Mucus penetration |
pSLN |
Doxorubicin |
Class 1 |
Caco-2/HT29 co-culture, Everted rat intestine, Intestine loops model, Healthy rat |
- -
-
↑ mucus penetration
- -
-
↑ bioavailability by 1.99-fold compared to non-PEGylated SLN
|
[56] |
| Mucoadhesive |
Chitosan coated liposome |
Alendronate |
Class 3 |
Caco-2 monolayer, Healthy rat |
- -
-
strong adsorption with mucins
- -
-
↑ bioavailability by 2.6-fold compared to alendronate solution
|
[57] |
| Enterocyte adhesive via WGA-lectin binding |
LPSN |
Paclitaxel |
Class 2 |
A549 cells, Healthy rats |
- -
-
↑ retention time in blood compared to free PTX
- -
-
↑ plasma AUC, peak concentration, t1/2 compared to free PTX
|
[58] |
| M-cell phagocytosis, TJ opening, and caveola-mediated endocytosis |
HACC-DTX-SLN |
Docetaxel |
Class 2 |
Caco-2 monolayer, FAE monolayer, Healthy rat |
- -
-
high Peyer’s patch accumulation
- -
-
Reversable regulation of tight junction
|
[59] |
| Lymphatic uptake |
NCC-SLN |
Curcumin |
CYP3A4 inhibitor |
Healthy rat |
- -
-
↑ plasma AUC by 9.5-fold compared to curcumin solution
- -
-
6.3-fold higher accumulation in lymph nodes than curcumin solution
|
[60] |
| pH responsive drug release (i.e., pH 1.2 and pH 7.4) |
EuC-NLS |
Alendronate sodium |
Class 3 |
Healthy rabbit |
- -
-
↑ bioavailability by 12-fold than ALS tablets without enteric coating polymer
|
[61] |
| pH-responsive drug release, (i.e., pH>7.0) |
IRSLNF3 |
Irinotecan hydrochloride trihydrate |
Class 1 |
Healthy mice, HT-29 bearing mice |
- -
-
↑ 1.62-fold in plasma AUC compared to NPs without coating pH sensitive microbeads
- -
-
↑ inhibition of tumor growth
|
[62] |
| Targeting MCT1 transport |
DTX-ACSL-Lip |
Docetaxel |
Class 2 |
4T1 and Caco-2 cells, Healthy rats |
- -
-
↑ 10.70-fold in plasma AUC compared to non-targeted DTX-lip
- -
-
GSH responsive release at tumor
|
[63] |
| Targeting ASBT in the distal ileum |
DSLN-CSG |
Docetaxel |
Class 2 |
Lymph fistula rat model, Healthy rats and tumor bearing mice |
- -
-
↑ bioavailability by 5-fold compared to non-modified NPs
- -
-
plasma DTX profile sustained up to 24 h
- -
-
enhanced tumor growth inhibition and prevention
|
[64] |
| Targeting VB12 mediated endocytosis |
H/VC-LPN |
Curcumin |
CYP3A4 inhibitor |
Caco-2/HT29-MTX co-culture, Healthy mice & rats |
- -
-
mucus penetration
- -
-
↑ plasma AUC by 13.89 folds and Cmax by 7.9 folds compared to curcumin suspension
- -
-
multiple pathways absorption, including M cells and transcytosis across epithelium
|
[65] |
3. Conclusions
In summary, there are many drug-drug interactions that occur due to the CYPs system. This usually should not pose a major issue because adverse drug reactions usually occur when drug concentrations are very high, very potent inhibitors are used, or when drugs are suicide substrates. However, drug-drug interactions are still occurring, especially in today’s society where people are engaging in polypharmacy. By designing orally administered LNS to address intestinal CYP3A4 metabolism, these strategies have the potential to improve bioavailability and reduce drug-drug interactions for susceptible CYP3A4 drug candidates. Pharmaceutical knowledge in the field of drug metabolism is important to reduce toxicity, reduce drug-drug interactions, and reduce the occurrence of beneficial drugs being removed from the market.
 +1 point
+1 point





