1000/1000
Hot
Most Recent
 +1 point
+1 point
Glioblastoma multiforme (GBM) is the most common primary brain cancer. GBMs commonly acquire resistance to standard-of-care therapies. Among the novel means to sensitize GBM to DNA-damaging therapies, a promising strategy is to combine them with inhibitors of the DNA damage repair (DDR) machinery, such as inhibitors for poly(ADP-ribose) polymerase (PARP).
Glioblastoma multiforme (GBM) has a radically altered genome and epigenome [1][2], which partially explains its aggressiveness. These modifications comprise point mutations, changes in the gene copy number, complete rearrangements, and epigenetic alterations [3]. Part of these genetic aberrations arise from the dysregulation of one or more molecular pathways responsible for recognizing and repairing DNA damage. Collectively, this molecular network is dubbed the DNA damage repair (DDR) machinery. When damaged DNA cannot be repaired, a functional DDR machinery triggers a signaling cascade, leading to cell senescence or apoptosis. These signaling pathways are disrupted in GBM, and despite the accumulation of DNA damage, cancerous cells will thrive, maintaining survival and pathological cell division [2][4].
While genomic instability contributes to the poor prognosis for GBM patients [5], the damaged DNA offers a target for pharmacological approaches that induce cancer cell death by a mechanism called synthetic lethality. This killing process occurs only if two molecular pathways are simultaneously deficient in one cell, whereas the isolated defect is innocuous [6]. One class of drugs that explores this killing mechanism are the inhibitors of poly(ADP-ribose) polymerase (PARP) enzyme, an important DDR component. PARP inhibitors (PARPis) can induce synthetic lethality in cancer cells with preexistent defects in the homologous recombination (HR) repair pathway, such as deleterious mutations of the breast cancer 1 (BRCA1) and breast cancer 2 (BRCA2) suppressor genes [7][8].
The majority of GBM clinical trials evaluating PARPis do not investigate the BRCA status to compare outcomes between patient groups [9]. One possible explanation is the expected low frequency of BRCA mutations in GBM, compared to breast, ovarian, and pancreatic cancer [10][11]. However, the reduction of BRCA expression due to the modulation of EF2 transcriptional factors, cyclin-dependent kinases changes in methylation of histones [12], or disruption of other DDR effectors [13] can similarly impair HR repair, resulting in the same phenotype observed for BRCA mutations. Collectively, these alterations that mimic BRCA mutations are known as ‘BRCAness’ [14]. Unraveling of GBM biology found mutations [1] in other genes that also result in BRCAness and confer sensitivity to PARPi treatment.
The increased understanding of GBM molecular landscape [1][15][16] has revealed promising biomarkers for prognostic assessment, resistance mechanisms [4][15], and targets for new therapies. We examine here the value of biomarkers that indicate the BRCAness status in GBM as an objective decision-making criterion for adding PARPi in the current glioma therapy.
PARP is a family of enzymes that comprises 17 members with different functions, such as maintenance of genomic stability, transcriptional regulation, and cell death [16][17]. PARP1 is the best-characterized member of this family and plays a pleiotropic role in DDR, thus becoming an attractive target for cancer therapy [18]. The major role of PARP1 is binding to and repairing DNA insults, mainly single-strand breaks (SSBs) through the base excision repair (BER) pathway [19]. PARP1 uses NAD+ as a substrate to modify acceptor proteins with poly(ADP-ribose) (PAR) and forms a scaffold, which in turn attracts BER components [16][17].
SSBs occur endogenously [20] and can evolve into double-strand breaks (DSBs) during DNA replication, which is the most deleterious DNA insult. In turn, DSBs are repaired by a complex network of proteins that belong to HR. BRCA1 and BRCA2 work at different stages of HR [15] and are required for a fool-proof repair of DNA DSBs through the HR pathway [12]. BRCA1 is a protein that functions in both checkpoint activation and DNA repair. Differently, BRCA2 mediates the core mechanism of HR [21]. HR happens during the S and G2 phases of the cell cycle due to the requirement for a sister template to promote DSB repair [22]. If there is a deleterious mutation of the BRCA1/2 genes, DSB will be fixed by the nonhomologous end-joining (NHEJ) pathway, which is an error-prone pathway. Therefore, BRCA1/2 mutated cancer cells are heavily reliant on a DDR repair pathway that will promptly fix SSBs before they become the deleterious DSBs [18]. Conversely, if PARP activity is inhibited in BRCA-deficient cancer cells, genomic aberrations accumulate beyond a bearable threshold and cause cancer cells to die by synthetic lethality, as depicted in Figure 1 [6][7][23].
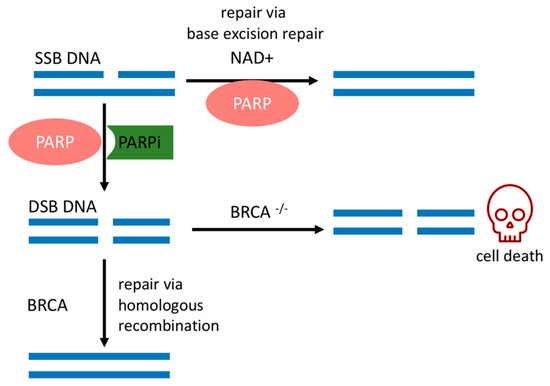
The sensitivity of BRCA mutated cancer cells to PARP1 inhibition has brought this enzyme to the forefront of therapeutic interest, and multiple potent candidate molecules have been and continue to be developed [7][8]. All PARPis approved for clinical use, or tested in clinical trials, consist of small synthetic molecules that interact with the NAD+ binding domain. Each PARPi presents a unique pharmacokinetic profile, efficacy [24], and cytotoxicity. Cytotoxicity is dependent on two concurrent mechanisms, namely (1) catalytic inhibition of PAR polymer formation and (2) trapping of PARP1 onto the DNA lesion, forming a complex of PARP1–PARPi–DNA [6]. Their trapping potency is the most relevant factor that leads to synthetic lethality [25], due to the collapse of the replication fork when it encounters the trapped PARP1–PARPi–DNA complexes where deleterious DSBs are formed and cause further cell cytotoxicity [25][26]. This mechanism surpasses the effects of killing cancer cells of only unrepaired SSBs due to the absence of PARP1 in the cell [25].
Missense mutations of IDH1/2 genes disrupt HR and affect 9% of GBM tumors [27]. These genes code for enzymes of the citric acid cycle that catalyze the conversion of isocitrate to alpha-ketoglutarate, as depicted in Figure 2 a [28]. In contrast, mutated IDH enzymes will convert alpha-ketoglutarate to 2-hydroxyglutarate, an oncometabolite that inhibits lysine-specific demethylase 4A/B (KDM4A/B) [29]. Both enzymes remove histone trimethylation, allowing DDR proteins, such as 53BP1 (P53 binding protein1), to access and repair DSB [30]. Notably, preclinical studies reveal that IDH1/2 mutations predict susceptibility to PARPi therapy [29][31]. Taken together, IDH mutations are expected to cause a BRCAness phenotype through disruption of HR and lead to sensitivity to PARPi therapy in GBM cells [31].
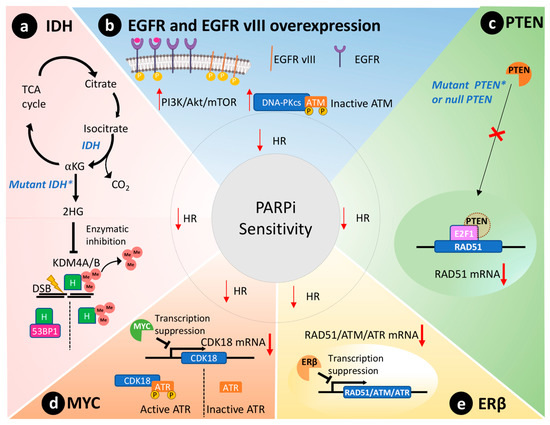
PTEN is silenced in 36% of GBMs [23]. Primarily, PTEN regulates the PI3K pathway, suppressing and having an important role in genomic integrity. Shen et al. demonstrated that PTEN through the transcription factor E2F1 transactivates the RAD51 gene promotor, ultimately controlling its gene transcription [32]. Cancer cells without functional nuclear PTEN, or lacking PTEN expression altogether, exhibit reduced RAD51 expression that leads to HR defects [33]. Figure 2 c schematically represents this molecular scenario. RAD51 is required to physically link homologous DNA molecules and a processed DNA break to catalyze the DNA strand exchange with an undamaged sister chromatid or homologous chromosome. Cancer cells lacking PTEN present less capacity to repair DNA insults caused by irradiation and are susceptible to PARPi therapy to the same extent as cancer cells presenting BRCA1/2 mutations [33]. Concurrently, PTEN-deficient glioma cells also presented an impaired HR and increased susceptibility towards PARPi therapy [34]. Lin and collaborators described that an orthotopic murine model of PTEN-deficient GBM was more susceptible to the association of veliparib and temozolomide than the control group with wild-type PTEN [35].
The MYC oncogene family encodes a set of nuclear phosphoprotein transcriptional factors that play key roles in cell cycle progression, apoptosis, cellular differentiation, and metabolism [36]. These genes were found amplified in a subset of GBMs by Hui et al. by genomic microarray analysis [37]. Increased overexpression/amplification of MYC was linked to increased sensitivity to PARPi therapy in patient-derived GSCs [13]. Cancer cells that overexpressed MYC also had a reduction in HR capacity and thus an increased susceptibility to PARPi [13]. Mechanistically, in GSCs, MYC downregulates CDK18, which in turn phosphorylates and consequently activates ataxia telangiectasia and Rad3-related (ATR) kinase, a key regulator of HR, thus conferring susceptibility to PARPi ( Figure 2 d) [13].
Estrogen receptors are proteins present in the cytoplasm and nucleus of cells where they modulate gene expression when they form a complex with estrogen hormone [38]. These receptors play a tumor-suppressive role in GBM models [39][40]. In agreement, aggressive GBM clinical samples showed reduced presence of ERβ in the nucleus compared to less aggressive gliomas and the normal brain tissue [41][42]. Additionally, the use of ERβ agonists increased the sensitivity of GBM to temozolomide treatment in preclinical models [40]. Transcriptome analysis of GBM cells overexpressing ERβ revealed mRNAs related to HR and DDR were downregulated, including RAD51, ATM, and ATR ( Figure 2 e) [43]. Hence, ERβ expression could be used as a biomarker to predict PARPi susceptibility in GBMs due to its role in modulating the expression of important DDR effectors and associated BRCAness phenotype.
GBMs are the most aggressive brain cancer, and they are known for genomic abnormalities and resistance to conventional therapies. A better understanding of the cancer biology of GBM has brought into attention the DDR cascade with the central role of PARP1 as a therapeutic target, opening up the opportunity for the use of PARPis in GBM therapy. This resulted in the continuous FDA approval of protocols using PARPis for a growing number of cancer types. Based on preclinical data, clinical trials are evaluating PARPis for the treatment of GBM patients. However, identification of consistent biomarkers that would allow predicting therapy response and elucidation of mechanisms of resistance to these drugs have so far failed.
Since PARP1 enzyme is the target of PARPi molecules, one important feature that must be assessed is its presence and the capacity of these drugs to engage with the enzyme throughout the treatment [44]. Currently, PARP1 levels are determined by immunohistochemistry in biopsy samples from GBM patients [44][45], but the known genetic and morphological heterogeneity of this cancer type limits this strategy as a diagnostic tool [1]. Furthermore, the engagement between PARP1 and its inhibitor requires that the PARPi molecules reach the cell nucleus and remain there to exert their therapeutic effect [46]. Therefore, a crucial characteristic of any PARPi is the capacity to accumulate in GBM tissue [47], evading the molecular efflux pumps [48].
Recent studies have shown that mechanisms of resistance to PARP inhibition are complex and multifaceted, including PARP1 mutations [49], CDK18-mediated resistance [13], and others [50]. Knowledge about these molecular processes and the heterogeneous genetic nature of GBM might explain why it is not possible to recapitulate what is observed in preclinical models in patients. Nonetheless, a growing number of clinical trials are now investigating biomarkers for PARPi sensitivity, and their results are awaited eagerly. Possibly, toxicities observed in some of the previous trials could be overcome with the use of more potent and more specific PARPis [25][51]. Ultimately, in future trials, with the advancement of biomarker research and the systematic use of ‘omics’ technology, a companion diagnostic for GBM should be developed, as has already been done for other cancer types. This will allow a better selection of patients and an optimal combination of targeted therapies.





