1000/1000
Hot
Most Recent
 +1 point
+1 point
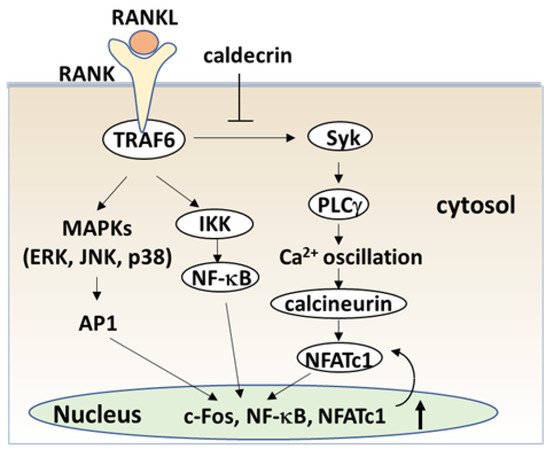
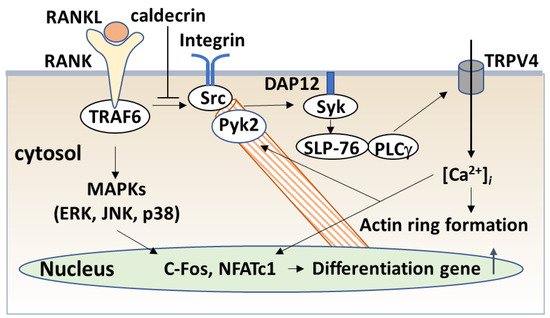
Blood calcium homeostasis is critical for biological function. Caldecrin, or chymotrypsin-like elastase, was originally identified in the pancreas as a serum calcium-decreasing factor. The serum calcium-decreasing activity of caldecrin requires the trypsin-mediated activation of the protein. Protease activity-deficient mature caldecrin can also reduce serum calcium concentration, indicating that structural processing is necessary for serum calcium-decreasing activity. Caldecrin suppresses the differentiation of bone-resorbing osteoclasts from bone marrow macrophages (BMMs) by inhibiting receptor activator of NF-κB ligand (RANKL)-induced nuclear factor of activated T-cell cytoplasmic 1 expression via the Syk–PLCγ–Ca2+ oscillation-calcineurin signaling pathway. It also suppresses mature osteoclastic bone resorption by RANKL-stimulated TRAF6–c-Src–Syk–calcium entry and actin ring formation. Caldecrin inhibits lipopolysaccharide (LPS)-induced osteoclast formation in RANKL-primed BMMs by inducing the NF-κB negative regulator A20.
Calcium homeostasis is controlled by calcium absorption in the intestine and reabsorption along the renal tubules, as well as by bone formation and resorption. Calcium homeostasis is regulated by parathyroid hormone (PTH) and thyroid gland-derived calcitonin, and activated vitamin D3 produced in the kidney. In addition, the pancreas is involved in calcium metabolism. Hypocalcemia is frequently observed in acute pancreatitis [1]. Glucagon [2][3], amylin [4][5], and calcitonin gene-related peptides [6][7] secreted from the pancreas may be responsible for hypocalcemia. While the exact pathological mechanisms of hypocalcemia are unknown, the pancreas may secrete hypocalcemic factors.
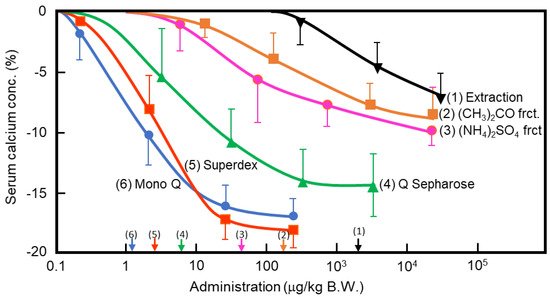
In the 1960s, Takaoka et al. first demonstrated that a pancreatic extract of porcine had hypocalcemic activity [8][9]. In 1992, Tomomura et al. purified a hypocalcemic factor, calcium-decreasing factor (caldecrin), from a porcine pancreatic extract [10]. Caldecrin was purified from acetone powder of porcine pancreas via fractionation with acetone (30–60%) and saturated ammonium sulfate (45–60%), followed by ion-exchange chromatography on Q Sepharose Fast–Flow (pH 5.5), gel filtration on Superdex 75 fast protein liquid chromatography (FPLC), and ion-exchange chromatography on Mono Q FPLC (pH 5.5). The isolated caldecrin was confirmed as a single peak by high-performance liquid chromatography. To identify caldecrin, each fraction of the purification steps was injected into the tail vein of fasted mice, and serum calcium concentrations were measured 4 h post-injection. The serum calcium concentration decreased in a dose-dependent manner as the products of subsequent purification steps were administered, and the hypocalcemic activity increased as a result of the purification process (Figure 1).
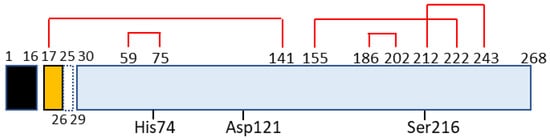
Chymotrypsin C, also termed caldecrin, is a 268-amino acid-long protein. Its sequence comprises a signal peptide (16 amino acids long, from residues 1 to 16), pro-peptide (13 amino acids, residues 17–29), and mature protein (239 amino acids, residues 30–268). The crystal structure of bovine Ctrc [15] revealed that rat and human caldecrin have five disulfide bridges: Cys17–Cys141, Cys59–Cys75, Cys155–Cys222, Cys186–Cys202, and Cys212–Cys243 [11][16] (Figure 2).
The enzyme classification of chymotrypsin C (caldecrin) is EC 3.4.21.2. It shows hydrolytic activity that can cleave leucyl, tyrosyl, phenylalanyl, methionyl, tryptophanyl, glutamine, and asparagine bonds. Chymotrypsin C preferentially hydrolyzes leucyl bonds compared to chymotrypsin A [17]. Humans have five other chymotrypsin-like elastase genes that encode the structurally similar proteins chymotrypsin-like elastase family, member 1 (CELA1, EC3.4.21.36), pancreatic CELA2A and 2B (EC3.4.21.71), and pancreatic CELA3A and 3B (EC3.4.21.70). The protease activity of CELA2 preferentially cleaves leucine, methionyl, and phenylalanyl residues and hydrolyzes elastin [18]. CELA3B preferentially cleaves alanyl residues, but has little elastolytic activity [19]. Human caldecrin was more similar to CELA2A (63.4%), 2B (59.6%), 3A (52.2%), and 3B (53.0%) than with chymotrypsin B (42.5%) [16].
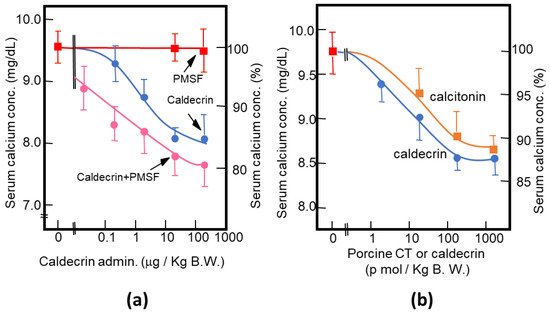
As described above, purified porcine and rat caldecrin from the pancreas and produced recombinant rat and human caldecrin protein decreased serum calcium concentration in mice. Caldecrin dose-dependently decreased the serum calcium concentration. The effect resulted in a maximum decrease of 15–20% with 20–100 μg (about 0.7–3.5 nmol)/kg mice body weight. Procaldecrin did not exhibit serum calcium-decreasing activity, but acquired serum calcium-decreasing and protease activity after trypsin treatment. PMSF treatment after the trypsin activation of procaldecrin abolished its protease activity but did not affect the serum calcium-decreasing activity [10] (Figure 3a). The calcium-decreasing activity of porcine caldecrin was almost the same as that of porcine calcitonin (1 nmol/kg body weight) (Figure 3b).
The serum calcium concentration is affected by osteoclast activity. Osteoclasts execute bone resorption, which is differentiated from bone marrow by key molecules such as macrophage colony-stimulating factor (M-CSF) and receptor activator of nuclear factor-kappa B (NF-κB) ligand (RANKL) [20][21][22][23][24]. Osteoclast differentiation and maturation occur in the following stages: (i) osteoclast precursor cells are produced from bone marrow cells in response to M-CSF and begin to differentiate following stimulation by RANKL; and (ii) osteoclasts fuse with each other to form multinucleated giant cells. Multinucleated cells secrete protons and cathepsin K, which are required for bone resorption. Osteoclast differentiation is tightly regulated by many molecules to maintain bone homeostasis [25][26][27]. (Figure 4).
Inflammation is known to cause bone loss. Bacterial lipopolysaccharide (LPS), a major constituent of the outer membrane of Gram-negative bacteria, is a potent inducer of bone loss in inflammatory diseases, including periodontal disease, bacterial arthritis, and dental implant infections [35][36]. Toll-like receptor (TLR) family members, which are proteins homologous to the Drosophila Toll protein, play a critical role in the innate immune system. TLR (TLR1–9) is expressed in osteoclast progenitors, of which TLR2 and 4 are also expressed in osteoclasts [37]. LPS has been shown to stimulate osteoclast formation and bone resorption in vivo through TLR4 [38][39].
The signaling cascade of TLR4 has been extensively studied in macrophages [40][41][42]. LPS induces inflammation upon the production of pro-inflammatory cytokines, such as interleukin-1 (IL-1) β, TNF-α, and IL-6 in macrophages and lymphocytes. Activated TLRs, except for TLR3, induce pro-inflammatory cytokine production through the canonical myeloid differentiation factor 88 (MyD88), which recruits TRAF6 downstream and activates IKK and the NF-κB pathway [43], leading to osteoclast formation in vitro. Although LPS is known to induce bone loss in vivo, LPS can both inhibit and stimulate osteoclastogenesis in vitro. The simultaneous activation of TLR4 and RANK signaling by LPS and RANKL, respectively, inhibits osteoclast formation in BMMs [37][44]. In this context, LPS/TLR4 activates NF-κB, p38, ERK1/2, and JNK, but inhibits RANKL-induced Nfatc1 expression. In contrast, LPS treatment enhanced the osteoclast differentiation of BMMs primed with M-CSF and RANKL [44][45]. The expression of Nfatc1 in RANKL-committed preosteoclasts is no longer affected by subsequent LPS treatment [45]. Therefore, RANKL-primed NFATc1 expression is a prerequisite for LPS-stimulated osteoclast formation.
RANKL/RANK and LPS/TLR4 signaling pathways in osteoclast formation share TRAF6, a ubiquitin E3 ligase, and downstream signaling pathways such as NF-κB activation. The LPS response is regulated by negative feedback with an NF-κB-inducible A20, which is a deubiquitinating protease encoding tumor necrosis factor alpha-induced protein 3 (TNFAIP3) [46]. A20 removes lysine 63 (K63)-linked polyubiquitin chains from TRAF6 and promotes K48-polyubiquitination for proteasomal degradation; thus, NF-κB-stimulated A20 plays an anti-inflammatory role by inhibiting IκB phosphorylation and NF-κB activation [47]. LPS induces osteoclast formation from RANKL-pretreated macrophages and the expression of A20, which is associated with TRAF6 degradation and NF-κB inhibition [48].
TREM2/DAP12 signaling contributes to macrophage activation. Tissue macrophages have two key functions: (1) to interact with pathogens such as LPS and modulate the adaptive immune responses, and (2) to facilitate tissue repair and tissue regeneration. These macrophage polarizations, categorized as M1 and M2, are modulated by the chemokine system [49][50][51]. Macrophages activated by LPS or interferon-γ alone or in combination are differentiated as classical M1 activation, which produces pro-inflammatory cytokines, whereas Th2-related cytokines IL-4 or IL-13, and anti-inflammatory molecules such as IL-10 and TGF-β, promote alternative M2 activation, which shows an anti-inflammatory and pro-healing phenotype [51][52]. TREM-2/DAP12 signaling contributes to the negative regulation of LPS/TLR4-mediated M1 macrophage polarization [53]. TREM-2-deficient macrophages enhanced the expression of pro-inflammatory cytokines and suppressed phagocytosis following TLR4 stimulation with LPS, demonstrating that TREM-2 suppresses inflammation and promotes bacterial clearance.
Rheumatoid arthritis (RA) is an autoimmune disease characterized by osteoclast-mediated bone and cartilage destruction resulting from inflammation in the synovium [54][55][56]. Osteoclast precursor cells are identified in areas of pannus invasion into the bone in RA. RANKL is expressed by both synovial fibroblasts and activated T lymphocytes derived from synovial tissues from patients with RA [57][58][59]. Pro-inflammatory cytokine levels, including TNF-α, IL1α, IL-1β, and IL-6, induce RANKL expression in synovial fibroblasts in RA, resulting in the enhancement of osteoclastogenesis in RA [60][61][62][63]. The A20 is decreased in monocytes and synovium from RA patients, suggesting that A20 may have a protective role in RA [64][65][66].
Recently, RANKL was reported to reduce muscular function when expressed in muscle cells. Anti-RANKL antibody treatment inhibits the NF-κB pathway and reduces muscle inflammation and damage in dystrophic mice [67]. Osteoprotegerin KO mice, which lack a secreted decoy receptor for RANKL, displayed reduced locomotor activity and signs of muscle weakness. Inhibiting RANKL improved the selective weakness and atrophy of fast-twitch IIb myofibers [68]. In addition, RANKL inhibition improved muscle strength and insulin sensitivity in osteoporotic mice and humans [69]. Therefore, caldecrin, by virtue of its anti-RANKL and anti-inflammatory activities, could be a suitable therapeutic approach for skeletal muscle dysfunction.





