Myeloid neoplasms encompass a very heterogeneous family of diseases characterized by the failure of the molecular mechanisms that ensure a balanced equilibrium between hematopoietic stem cells (HSCs) self-renewal and the proper production of differentiated cells. The origin of the driver mutations leading to preleukemia can be traced back to HSC/progenitor cells. Many properties typical to normal HSCs are exploited by leukemic stem cells (LSCs) to their advantage, leading to the emergence of a clonal population that can eventually progress to leukemia with variable latency and evolution. In fact, different subclones might in turn develop from the original malignant clone through accumulation of additional mutations, increasing their competitive fitness. This process ultimately leads to a complex cancer architecture where a mosaic of cellular clones—each carrying a unique set of mutations—coexists. The repertoire of genes whose mutations contribute to the progression toward leukemogenesis is broad. It encompasses genes involved in different cellular processes, including transcriptional regulation, epigenetics (DNA and histones modifications), DNA damage signaling and repair, chromosome segregation and replication (cohesin complex), RNA splicing, and signal transduction. Among these many players, transcription factors, RNA splicing proteins, and deubiquitinating enzymes are emerging as potential targets for therapeutic intervention.
1. Introduction
Hematopoietic stem cells (HSCs) ensure continuous production of all blood cell types throughout life. By means of a delicate equilibrium between self-renewal, quiescence, and differentiation, HSCs maintain homeostatic conditions and dynamically respond to stress stimuli. The balanced production of the different cell lineages physiologically varies during aging, when the hematopoietic potential is progressively skewed towards the myeloid lineages at the expenses of immune cells [1][2][3]. The accumulation of heritable genetic mutations in individual cells and the kinetics of their selection can lead to myeloid neoplasms, a heterogeneous group of diseases characterized by the dysfunctional production of myeloid cells in the bone marrow. This can manifest in cytopenia and cellular dysplasia, such as in myelodysplastic syndromes (MDSs), in the overproduction of mature clonal myeloid elements, such as in myeloproliferative neoplasms (MPN), or both, as it happens in MDS/MPN, which share different molecular and clinical traits and present both myelodysplastic and proliferative features [4]. Myeloid neoplasms entail a high risk of developing into acute myeloid leukemia (secondary AML or s-AML). AML can also develop as a late complication in patients after leukemogenic therapies (therapy-related AML or t-AML), or without clinical history of prior MDS or known exposure to potentially leukemogenic agents (de novo AML) [5]. Driver mutations leading to a preleukemia condition originate in HSC or in hematopoietic stem-progenitor cells (HSPCs). The preleukemic clones can have a variable latency and, in some cases, can persist for years before further mutations trigger their leukemic evolution [4][6].
MDS has an estimated crude incidence of 4 to 5 cases per 100,000 persons per year. Although MDS occurs at all ages, the incidence is higher in elderly individuals, with a median age of 70 years old at diagnosis [4]. Careful evaluation of the individual patient prognostic risk, genetics, and age guides the clinical decision-making process [4]. Although current drugs can temporarily modulate myelodysplastic hematopoiesis, they fail in eradicating the disease [4][7].
Failure of current therapies in eradicating MDS/AML is attributed to the persistence of leukemic stem cells (LSCs) upon treatment and to the emergence of resistant subclones [8]. Notably, recent deep-sequencing studies revealed the possibility that relapse from chemotherapy can occur not only from LSCs that are resistant to the treatment but also from pre-leukemic mutated but not transformed HSCs, which are already present in the patient at diagnosis or during clinical remission, and which can acquire additional mutations, further highlighting the complexity and heterogeneity of the disease [9][10]. LSCs share functional properties with normal HSCs [11]. Indeed, LSCs are functionally defined as cells capable of self-renewal and of propagating the disease upon transplantation into immunodeficient mice [8]. In addition to self-renewal capacity, LSCs co-opt many survival mechanisms typical to HSCs to their advantage, including genome maintenance processes, epigenetic and stemness transcriptional programs, the pre-mRNA splicing machinery, metabolic properties, interaction with the microenvironment, and inflammatory signals [11][12]. In fact, recurring mutations in regulators of gene expression, including epigenetic proteins, transcription factors (TFs), and the components of the splicing machinery, are found in 70% of AML patients [13][14]. Understanding the molecular mechanisms regulating HSC biology and their dysregulation in pre-leukemic HSCs and in LCSs is therefore critical for understanding the disease and for developing therapies that harness cancer HSPC-specific vulnerabilities and ultimately eradicate the disease.
2. Emerging Therapeutic Targets
2.1. Transcription Factors’ Targeting
In principle, depending on the nature of the oncogenic mutation, several approaches for therapeutic targeting of TFs could be envisaged in order to hit the oncogenic protein, or to restore the correct level of expression of the wild-type protein. The final goal is to limit the expansion of HPSCs and to restore the proper differentiation pathways (“differentiation therapy”). The best proof of principle of the efficacy of this differentiation approach is demonstrated by the successful use of ATRA (all trans retinoic acid) plus arsenic-trioxide (ATO) in the treatment of promyelocytic leukemia caused by the PML/RARa translocation, which made this disease curable
[15][16].
However, targeting TFs is very difficult because of the absence of enzymatic activity and of evident druggable domains. Despite this, a variety of new targeting strategies are under development. They target the aberrant oncogenic proteins or their specific interactions, the recruited epigenetic cofactors, the TF–DNA interaction, or the TF expression level (Figure 1).
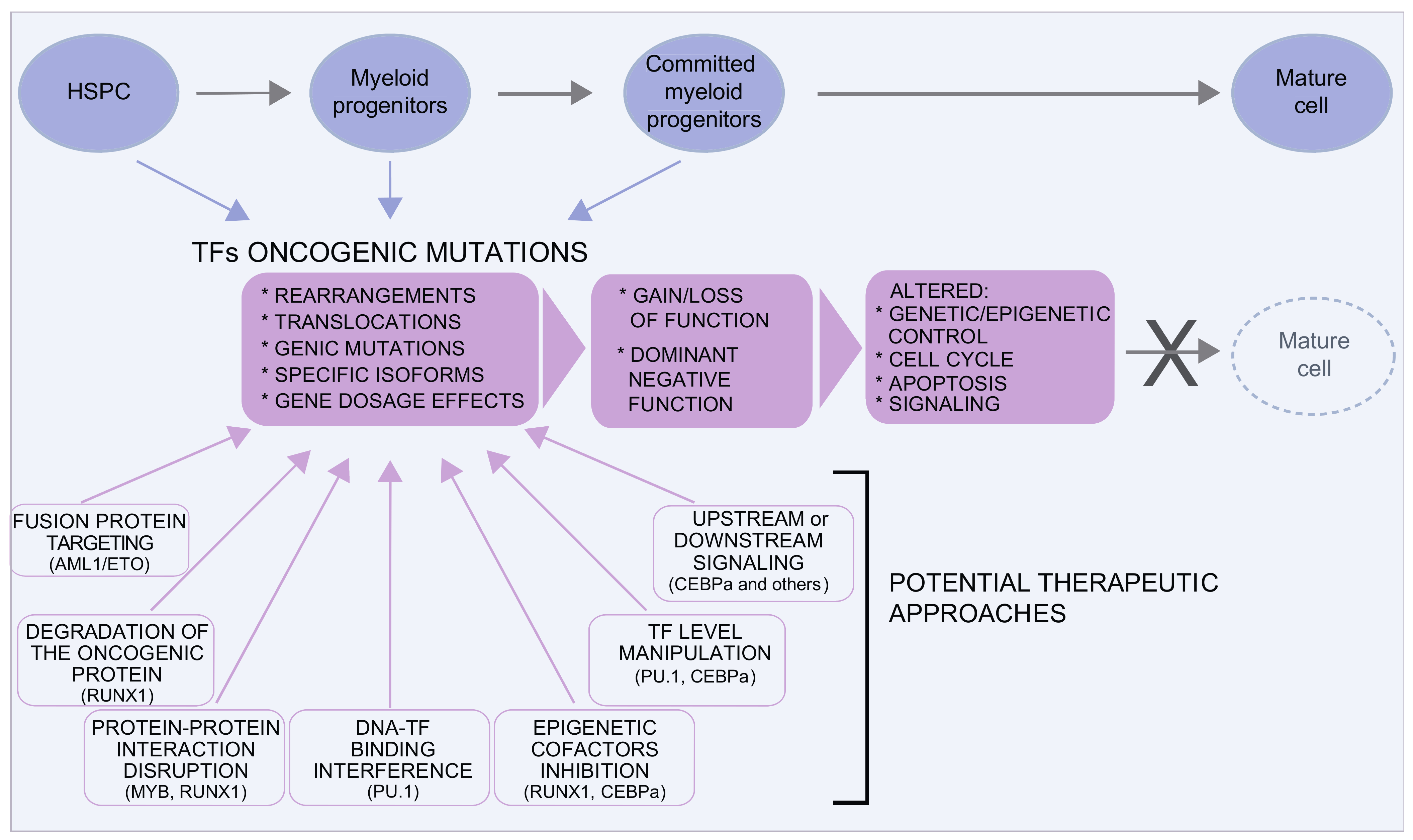 Figure 1.
Figure 1. Transcription factors in MDS/AML. Several transcription factors are involved in MDS and AML. Oncogenic mutations are very heterogeneous and can result in gain or loss of function or in dominant negative effects, often with a pleiotropic effect on the control of the equilibrium between proliferation and differentiation. Although traditionally considered “undruggable” factors, in the last years, several approaches have been envisaged to either target the oncogenic protein or to restore the level/function of the TF. Some examples in this direction are discussed in the text and illustrated here.
2.2. Targeting the Spliceosome
Hotspot SF mutations are invariably heterozygous, indicating that cells rely on the expression of the wild-type allele for survival. This observation suggests that mutant cells could have increased sensitivity to pharmacological perturbation of the spliceosome by splicing modulator drugs. In recent years several naturally derived and structurally distinct splicing inhibitors/modulators (e.g., pladienolides, herboxidienes, and spliceostatins, reviewed in
[17]) have been identified. Many of these compounds target SF3B1
[18][19]. Notably, the K1071, R1074, and V1078 mutations confer resistance to these compounds
[20]. Recent studies of the cryoEM structures of the SF3b complex bound to spliceosome modulators have shown that they primarily bind to the adenosine branch point binding pocket
[21][22]. Artificial derivatives of these natural compounds have been developed that are currently under consideration in preclinical models of leukemia with SF mutations and in Phase I clinical studies. For example, Shirai and colleagues showed that
U2AF1(
S34F)-expressing hematopoietic cells are sensitive to sudemycin D6
[23], a small-molecule spliceosome inhibitor
[24] that also targets SF3B1
[25]. Treatment of
U2af1(S34F) transgenic mice with sudemycin results in an attenuation of mutant
U2af1-induced hematopoietic progenitor cell expansion that is associated with increased cell death
[26]. More recently, H3B-8800, a pladienolide-derived artificial small molecule that induces antitumor activity in xenograft leukemia models with core spliceosome mutations
[27] has been used in a Phase I clinical trial (ClinicalTrials.gov Identifier: NCT02841540) in patients with MDS, AML, or in chronic myelomonocytic leukemia (CMML)
[28][29]. In addition to SF3B inhibitors, very recent work has identified phenothiazine derivatives that inhibit early spliceosome assembly by specifically inhibiting the interaction of U2AF homology motifs (UHM) with U2AF ligand motifs (ULMs)
[30]. These drugs could potentially interfere not only with the heterodimerization of the U2AF1 and U2AF2 but also with the activity of other splicing factors that contain UHM domains.
However, due to the fundamental role that pre-mRNA splicing plays in gene expression, there are concerns about the potential widespread toxicity of spliceosome inhibitors. Indeed, a Phase I clinical trial using E7107, a pladienolide derivative, against solid tumors was halted after two participants developed vision loss as a potential side effect of the drug
[31].
2.3. Are DUBs Possible Therapeutic Targets?
Deubiquitinating enzymes are a relatively new class of drug targets. DUBs contribute to cancer through, at least in part, stabilization of the oncogenic proteins, modulation of the activity of the oncogenes or tumor suppressors proteins, or control of the epigenetic changes that promote tumor development
[32]. Therefore, the generation of small-molecule inhibitors of DUBs is currently an active pursuit of the pharmacology industry
[33][34]. Despite the intensive pre-clinical studies on selected DUBs, clinical translation of DUB inhibitors remains challenging and no inhibitor is currently being tested in active clinical trials
[33][34].
3. Conclusions
The advent of extensive sequencing technologies has offered an unprecedented opportunity to gain insight into the complexity of the mutational landscape underlying myeloid neoplasms and their evolution, allowing to progressively define and refine molecular sub-groups.
In parallel with the expansion of sequencing technologies, the better understanding of the molecular mechanisms governing HSPCs biology and the lineage choice leading to the balanced production of mature cells has provided the rationale for the identification of novel potential targets/drugs for therapeutic interventions, as in the case of transcription factors, splicing factors, and deubiquitinating enzymes, the new key players in MDS and AML discussed in this review.
The paradigm of TFs being “undruggable” molecules is now challenged by a variety of targeting approaches. Small molecules inhibiting but also activating TFs are available. “Epigenetic therapy” directed against the chromatin modifiers recruited by TFs possesses great potential for wide application, but, for this same reason, raises serious specificity concerns. More recently, the direct targeting of specific protein–protein or DNA–TF interactions, by either masking the DNA-binding consensus on the DNA or by blocking the DNA-binding domain of the TF, have been proposed. Finally, the degradation of oncogenic TFs by the destabilization of their complexes, by miRNA-mediated degradation or by tagging them with ubiquitin, is becoming, in principle, an option. The targeted degradation approach could also be useful when a low level of a TF promotes cancer cell expansion but its further reduction induces cancer cell death.
The rational underlying modulation of pre-mRNA splicing in MDS carrying splicing factors’ mutations is to induce synthetic lethality. In this respect, small-molecule spliceosome inhibitors have demonstrated a high potential as novel therapies. However, since these drugs target the core components of the splicing apparatus, concerns about toxicity need to be addressed in pre-clinical and clinical trials. Nevertheless, inhibitors of additional splicing targets are in development and more therapeutic approaches will likely be determined. For example, R-loop formation, which is increased by splicing factors’ mutations, can sensitize cells to ATR inhibition.
In recent years, DUBs have been clearly confirmed to affect normal and malignant HSCs. Cellular and in vivo studies indicate that the functional and/or pharmacological inhibition of DUBs has therapeutic benefit in MDS/AML. Although the complexity of the DUB domains and the conservation of catalytic pockets pose challenges to the clinical development of small-molecule DUB inhibitors, recent X-ray crystallographic studies have demonstrated the possibility of designing selective and potent inhibitors with antitumor activity, as exemplified by the USP7 inhibitors that have therapeutic effects in AML in vivo models. The efficacy of co-application of DUB inhibitors with chemotherapy or kinase inhibitors, as well as their application to promote degradation of (onco) proteins with inherent or acquired resistance are valuable directions of research. As multiple DUBs contribute to HSC dysregulation in leukemogenesis, an in-depth understanding of their biological role and complex interactions in HSC biology and their perturbation in pre-malignant HSCs and LSCs is essential to the design of novel combinatorial treatments that affect the self-renewal of LSCs while sparing normal HSCs.
Large-scale population screenings have revealed that very often mutations found in AML cells are also present in non-malignant cells, in particular in elderly subjects. This observation poses the problem of evaluating the functional significance of this mutational heterogeneity in terms of the leukemic potential and functional relationship (cooperation, co-inhibition or neutrality) of co-occurring mutations.
In this scenario, molecular genetic screening at diagnosis, during follow-up for minimal residual disease monitoring, and at relapse will be crucial to trace the in vivo evolution of premalignant and malignant clones, with important implications for the prognosis and for the design of appropriate targeted therapies. Parallel functional approaches in cellular and patient-derived xenograft models will be complementary to assess the functional relevance of the mutations and to identify critical common mutations and/or pathways that could overcome the genetic heterogeneity and the mutational complexity of cancer cells. Hitting such common targets concomitantly with specific gene mutations through the design of combination treatments is a crucial issue for the development of effective therapies.
Although this new knowledge on key players and on the genetics and cellular aspects of leukemia onset and evolution have not yet reached full clinical translation, these studies will contribute to increase the repertoire of possible strategies to control, if not to eradicate, the disease through a personalized medicine approach.
 +1 point
+1 point





