3. Melatonin and Chronic Pain
Pain has been defined as “An unpleasant sensory and emotional experience associated with actual or potential tissue damage or described in terms of such damage”
[33]. Pain significantly affects the quality of life and implies increased social costs for its management
[34]. A relationship between pain and melatonin has beendelineated, since chronic pain patients have shown lower levels of melatonin in the blood and urine
[35]. Even the precursors of melatonin such as L-tryptophan and serotonin were found to be low in patients with fibromyalgia, indicating that the melatonin has a critical role in chronic pain syndromes
[35][36]. Further adding to the evidence, analgesia observed after melatonin administration in both nociceptive and neuropathic pain models also points towards an important role of melatonin in regulating pain
[37]. Melatonin affords amelioration of pain through several mechanisms. For instance, Melatonin could ameliorate the nociceptive pain owing to its ability to prevent prostaglandins release, inhibition of the migration of polymorphonuclear cells at the inflammation site, and inhibition of the cyclooxygenase-2 (COX-2) and nitric oxide (NO) synthase. Melatonin provides neuropathic pain relief through its ability to raise the pain threshold and decrease of the thermal hyperalgesia manifestation. Intriguingly, the analgesic effects of melatonin have been shown to be dose dependent and have been observed consistently across various pain induction models relying on thermal, chemical, mechanical and electrical pain induction
[37]. Furthermore, the different routes of administration almost provide the same efficacy from a given dose of the melatonin
[37]. Analgesic effects of melatonin have been well appreciated across various chronic pain syndromes such as chronic pelvic pain, fibromyalgia, irritable bowel syndrome, tension and cluster headaches, migraine and others including chronic back pain
[37][38]. Around 5–10% of the patients with acute back pain progress to subacute back pain category and then, finally develop chronic back pain, which is defined as back pain lasting for more than 3 months
[34]. The next section will provide an overview of the melatonin’s pain-relieving mechanisms before discussing the melatonin’s effects in chronic pain disorders of different origin.
3.1. Mechanisms of Melatonin Action in Relieving Pain
The pain-relieving effects of melatonin either stem from its direct anti-nociceptive effects due to its anti-oxidant, anti-inflammatory properties and direct interaction with its cognate receptors or through indirect complex interactions with other systems that can modulate the pain perception pathways
[29][37].
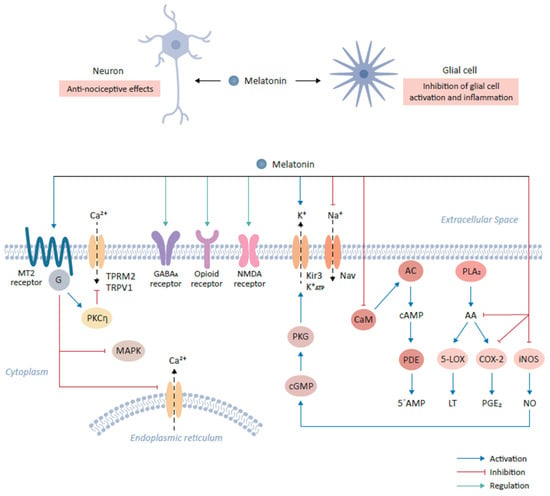
MT2 receptors have been found to be particularly enriched in the brain areas which play an important role in modulating the pain sensation
[29]. For example, ventromedial nucleus of the thalamus and reticular formation as a part of the ascending nociceptive pathway and ventrolateral periaqueductal gray matter as a part of the descending antinociceptive pathway harbor high densities of the MT2 receptors
[32]. Initial studies involving the use of melatonin antagonists also confirmed that MT2 receptors are preferentially implicated in mediating melatonin’s anti-nociceptive effects
[32]. Furthermore, selective partial agonists at MT2 receptors such as UCM765 and UCM924 have shown anti-nociceptive effects in experimental models of nociceptive pain
[29][37]. Modulation of the MT2 receptors expressed on glutamatergic neurons in the rostral ventrolateral periaqueductal gray by UCM924 decreased tail flick responses in experimental pain model and modulated the neuronal activity of the pain pathways
[29][37]. An agonist of MT2 receptor, IIK-7, provides relief against neuropathic pain via inhibition of glial cells activation and inhibition of inflammation as exhibited by down regulation of inducible nitric oxide synthase (iNOS) and caspase 3
[29]. Intriguingly, MT2 receptor ligation by melatonin reduces neuronal excitability in experimental pain model owing to inhibition of T-type Ca
2+ channel currents via downstream G
βγ-mediated PKCη signal pathway (
Figure 1). In addition, melatonin suppresses mechanical allodynia and thermal hyperalgesia due to the inhibition of the mitogen activated protein kinase (MAPK) and calcium signaling
[29].
3.2. Interaction with Neurotransmitters (and Their Receptors)
Melatonin interacts with various neurotransmitter systems indirectly to produce nociceptive effects. For instance, melatonin can produce antinociceptive effects by affecting GABAergic (GABA
A receptor), benzodiazepinergic (BZD receptor), opioidergic (δ/µ/κ receptors), sigma system (sigma receptor), dopaminergic receptor (D
2 receptor), adrenergic (α
2 receptor), serotonergic (5-HT
2A receptor), glutamatergic (NMDA receptor) and NO-cyclic GMP-PKG signaling pathway (
Figure 1)
[30].
Melatonin can modulate the GABAergic transmission as was evidenced from the observation that Flumazenil, which is a benzodiazepine antagonist and inhibits indirectly the GABAergic neurotransmission via antagonizing the allosteric effects of benzodiazepines receptors on their own receptor subtypes and therefore reversing the anti-nociceptive effects of Melatonin
[32][37][38][39]. Melatonin is known to facilitate GABAergic transmission through various mechanisms such as the increase of the GABA content, increased affinity of GABA for its receptors and density of GABA receptors; even melatonin and its analogues can directly bind to GABA receptors
[31][32][37][38][39]. These mechanisms likely explain the synergistic effects of melatonin and benzodiazepine-GABA agonists, which are independent from direct interaction of melatonin to its cognate receptors (
Figure 1).
Interestingly, there is evidence that there exists an indirect link between melatonin and the opioidergic system. For instance, the melatonin’s antinociceptive effects were antagonized upon Naloxone administration, which is an opiate antagonist
[37][38]. Further, melatonin administration could induce the production and release of β-endorphins; and morphine and other opioid agonists could induce the melatonin release from the pineal gland
[39]. Furthermore, the analgesic effects of morphine were reduced in pinealectomized mice. Intriguingly, neither melatonin nor any of its analogues were shown to directly interact with opioid receptors, yet there exists an important connection between melatonin and opioids in mediating anti-nociceptive effects
[37][38][39].
Glutamate and its NMDA receptors have been implicated in nociceptive sensation pathways
[29]. Repetitive firing of C-fibers potentiates the spinal nociceptive transmission, an effect known as wind-up, and is dependent on NMDA receptors. Interestingly, melatonin can induce suppression of this wind-up effect in a dose dependent manner through the modulation of intracellular NO levels leading to the activation of cGMP-PKG-ATP sensitive K
+ channels
[30]. The antinociceptive effects of melatonin involve modulation of the α
1-adrenergic, α
2-adrenergic, muscarinic and nicotinic receptors at the spinal level through the modulation of intracellular cGMP levels (
Figure 1)
[30].
3.3. Modulation of Ion Channel Activity
Melatonin has been shown to activate the G-protein coupled Kir3 (inwardly rectifying K
+) channels at the cellular level, which reduce the rapid firing of action potential trains in neurons
[37][38]. Melatonin activates outward flow of K
+ ions in various areas of the CNS such as cerebellum, suprachiasmatic nucleus , while in turn it inhibits the activity of voltage gated sodium channels such as Nav1.8 and Nav1.9 to exert anti-thermal hypersensitivity and anti-mechanical allodynia effects
[29][37][38][39]. In the cultured dorsal root ganglion cells of the spinal cord, melatonin has been shown to decrease the neuronal free intracellular Ca
2+ levels via inhibition of the voltage sensitive Ca
2+ channels
[37][38][39]. This effect of melatonin to inhibit mobilization of free Ca
2+ from intracellular storage sites as well as inhibition of inward flow of Ca
2+ from voltage activated channels has also been demonstrated in various other tissues
[29]. Melatonin regulates against the Ca
2+ influx through the desensitization of transient receptor potential vanilloid type 1 (TRPV1) and transient receptor potential melastatin type 2 (TRPM2) channels (
Figure 1)
[29].
Melatonin is known to bind calmodulin and directly affect the calcium signaling and modulate the activity of the enzymes such as adenylate cyclase and phosphodiesterase or structural proteins
[28].
It is well established that Ca
2+ channels play a vital role in the development and maintenance of central sensitization that is associated with inflammation and neuropathic pain, melatonin, therefore provides pain relief through modulation of the activity of Ca
2+ channels
[37][38].
3.4. Melatonin and Inflammatory Mediators
The inflammation is upregulated by various lipid mediators generated from arachidonic acid by the action of Phospholipase A
2 (PLA
2), and subsequent activity of 5-Lipoxygenase (5-LOX) and Cyclooxygenase-2 (COX-2). Melatonin can inhibit arachidonic acid formation, acting as a negative regulator of PLA
2, inhibit the expression of 5-LOX and COX-2 leading to decreased synthesis of chemoattractant leukotrienes and prostaglandins, respectively
[37][39]. Melatonin is also known to decrease the expression of iNOS (inducible nitric oxide synthase) and COX-2, which subsequently leads to a decline in NO and PGE
2 expression, respectively
[39]. These inflammatory mediators are involved in inflammatory pain perception and therefore, the inhibition of these inflammatory mediators contribute to the analgesic effects of melatonin (
Figure 1).
4. Melatonin at the Crossroads of Different Chronic Pain Disorders, Sleep and Inflammation
Back pain is a commonly encountered musculoskeletal disorder that is often perceived to be related to disability. Most of the patients with acute back pain have good recovery, however, a small portion of the patients suffer from this pain over an extended period of time than acute back pain (more than 12 weeks) and are referred to have chronic back pain. Since, the incidence of acute back pain is higher, consequently the number of patients with chronic back pain is also getting higher
[40]. It is estimated that 30–60% of the population in developed countries suffer from chronic back pain and it affects mostly the people of working age with a peak prevalence in the age group of 30–60 years. This represents a high cost to the society and contributes to disability
[37][40]. It is of note that chronic back pain is responsible for the most years lived with disability when compared to any other medical illness
[40].
Furthermore, melatonin provides pain relief in several other chronic pain conditions such as migraine, chronic cluster and tension type headaches, fibromyalgia, irritable bowel syndrome and chronic pelvic pain. Intriguingly, melatonin administration to chronic back pain patients has been shown to significantly relieve the pain both at rest and during motion
[37][38]. In a clinical study, the addition of melatonin at a dosage of 3 mg/day, 30–40 min before sleeping at night, to the standard therapy consisting of administering glucosamine hydrochloride and chondroitin sulfate alone or in combination with diclofenac or only diclofenac was found to significantly reduce the pain intensity during movement and at rest as assessed by Visual Analog Scale (VAS) and related scoring batteries
[37][38]. Melatonin addition to the standard regimens also provided additional benefits such as reduced pain-associated anxiety, mood stabilization and sleep quality improvement, which are often impaired in the chronic back pain patients
[37]. These beneficial effects of melatonin might be due to the restoration of the circadian rhythm disturbed by the chronic pain, improvement in sleep and return of the body’s adaptive potential to normal besides the intrinsic analgesic properties of the melatonin administration. Intriguingly, in this study, the analgesic effects of melatonin were observed earlier than the sleep normalization effects, supporting the notion that the melatonin has its own intrinsic antinociceptive effects apart from similar effects brought about through complex interactions with other systems
[37].
On one hand, the pain substantially affects the sleep and on the other hand, deprivation of restful sleep escalates the pain susceptibility
[41]. So, both conditions can aggravate each other suggesting a bidirectional and reciprocal relationship, further adding to the suffering of the patient
[42]. Chronic pain can result in sleep deprivation for extended periods of time and can affect the physical, emotional and behavioral wellbeing of the patients
[41]. It is estimated that around 50–80% of the patients with chronic pain experience sleep disturbances
[42]. Interestingly, a study involving chronic back pain patients showed that 53% of the patients met the criteria for insomnia compared to 3% of the healthy pain free controls
[42]. Additionally, the severity of insomnia showed a positive association with pain intensity, sensory pain ratings, affective pain ratings, general anxiety, health anxiety and general depression. Clinical studies have shown that chronic pain patients reporting sleep disturbance have higher pain perception, more fatigue, poor mood, and increased levels of stress and disability. Alternatively, healthy subjects who were deprived of sleep showed an increased pain probably due to an increase in the release of pro-inflammatory cytokines and reduced pain tolerance
[42].
Melatonin is shown to improve the quality of sleep in chronic inflammatory pain patients and other chronic pain disorders in addition to its analgesic effects
[29][37]. Repeated administration of melatonin progressively improves the sleep and reduces anxiety, thereby leading to lower levels of pain
[43]. Intriguingly, in an experimental chronic constriction injury rat model, deprivation of sleep was shown to further aggravate the neuropathic pain and was associated with increased activation of microglia and lower levels of serum melatonin
[44]. However, when melatonin was administered, it attenuated the microglial activation, reduced the levels of pro-inflammatory cytokines ultimately leading to a reduction in neuropathic pain
[44]. A study involving the administration of melatonin to chronic back pain patients also showed a significant improvement in the sleep quality in addition to a reduction in back pain
[45].
A variety of inflammatory mediators are liberated upon tissue damage or inflammation including prostaglandins (PGE
2), leukotrienes, bradykinin, substance P and inflammatory cytokines, which either directly activate nociceptors or release local allogenic agents capable of further sensitizing the nociceptors leading to enhanced pain transmission
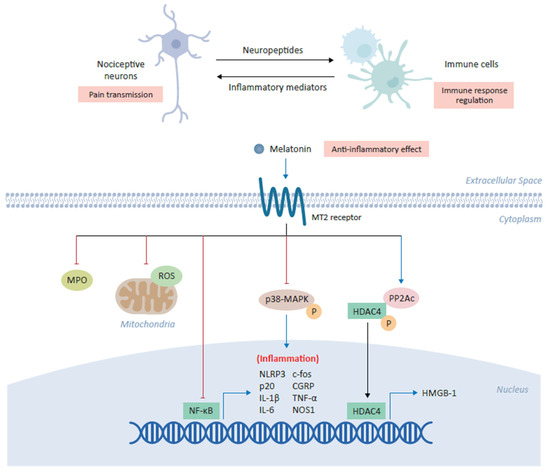
[30][46]. These inflammatory mediators setting the nociceptive system on fire are often produced by the immune cells. The coordination between nociceptive neuronal pathways and immune response represents a potential advantage to the host in protecting against danger (
Figure 2). Interestingly, neuroimmune communication in pain is bidirectional, for instance immune cell secreted mediators sensitize peripheral and central pain pathways and in turn, nociceptive neurons actively release neuropeptides via their peripheral nerve terminals to shape the response of innate and adaptive immune cells
[46].
Figure 2. Anti-inflammatory role of melatonin in alleviating chronic pain. Melatonin plays a central role in the coordination between nociceptive neurons and immune cells in which its binding the cross-talk between inflammation and pain transmission. The binding of melatonin to MT2 receptors expressed on immune cells inhibits myeloperoxidase (MPO) activity and NF-ĸB, as well as downregulating the production of mitochondrial reactive oxygen species (ROS). Melatonin has also been shown to inhibit the NLRP3 (NOD-, LRR- and pyrin domain-containing protein 3) inflammasome and modulate the downstream expression of several inflammatory genes, including c-fos, calcitonin-gene-related peptide (CGRP), IL-1β, IL-6, TNF-α and NOS1. In various cell types, melatonin inhibits the phosphorylation of p38 mitogen-activated protein kinase (MAPK). Finally, melatonin increases expression of the catalytic subunit of phosphatase 2A (PP2Ac) which in turn mediates the dephosphorylation of HDAC4, driving its nuclear accumulation to repress the expression of high mobility group box-1 (HMGB-1) and ultimately alleviating chronic pain.
Accumulating evidence suggests that melatonin can modulate the activation of immune system leading to a reduction in acute and chronic inflammation
[3]. Melatonin demonstrates anti-inflammatory effects through the inhibition of the inflammatory cells activation by inhibiting myeloperoxidase activity
[47]. Melatonin can produce anti-inflammatory effects through direct interaction with binding sites on macrophages and lymphocytes
[39]. Melatonin inhibits the release of pro-inflammatory cytokines and downregulates the expression of NFκB in various experimental studies. Consequently, melatonin administration was found to inhibit the release of TNF-α from peritoneal macrophages obtained from mice (
Figure 2)
[39]. Melatonin has been shown to inhibit NLRP3 (nucleotide-binding oligomerization domain, leucine-rich-containing family, pyrin domain-containing-3) inflammasome activation under various conditions
[47]. Interestingly, in an intervertebral disc degeneration (IVDD) rat model of low back pain, melatonin administration prevented IVDD progression and IVDD associated back pain, which was found to result from melatonin induced decrease in NLRP3, p20, and IL-1β levels due to inhibition of NFκB signaling and down modulation of mitochondrial ROS (reactive oxygen species) production
[48]. In another neuropathic pain study, it was revealed that melatonin via MT2 receptors in the DRG (Dorsal root ganglion) could alleviate the allodynia and hyperalgesia in a sciatic nerve cuffing model of mice (
Figure 2)
[36]. Interestingly, melatonin inhibited the activation of peptogenic neurons and neuroinflammation in DRG through downregulation of inflammatory genes such as c-fos, calcitonin-gene related peptide (CGRP), TNF-α, IL-1β and nitric oxide synthase 1 (NOS1)
[36].
In an Oxaliplatin induced neuropathic pain model, melatonin administration attenuated the astrocyte mediated spinal neuroinflammation and consequently, reduced pain hypersensitivity
[49]. Another study found that melatonin administration significantly reduced the neuropathic pain in a chronic constrictive injury with sleep deprivation experimental model and exhibited significant inhibition of the microglial activation along with lower levels of inflammatory cytokines such as TNF-α, IL-1β and IL-6 (
Figure 2)
[44]. Interestingly, chronic constrictive injury of the median nerve in the cuneate nucleus led to the phosphorylation of p38 mitogen activated protein kinase (p38-MAPK) and microglial activation, which was associated with behavioral hypersensitivity
[50]. Administration of melatonin reduced behavioral hypersensitivity, phosphorylation of p38-MAPK in microglia and consequently, reduced inflammatory cytokines secretion
[50]. Intriguingly, a spinal nerve ligation (SNL) pain model was used to unveil the melatonin’s underlying epigenetic mechanisms of chronic pain alleviation by controlling the inflammatory genes expression
[51]. SNL resulted in the reduced expression of the catalytic subunit of phosphatase 2A (PP2Ac) and increased histone deacetylase 4 (HDAC4) phosphorylation and cytoplasmic accumulation, which epigenetically relieved HDAC4-suppressed
hmgb1 gene transcription, resulting in enhanced high mobility group box-1 (HMGB-1) expression in the ipsilateral dorsal horn of the rats (
Figure 2). Notably, melatonin acting via MT2 receptors upon intrathecal administration increased PP2Ac expression, HDAC4 dephosphorylation and nuclear accumulation, restored the HDCA4 mediated suppression of
hmgb1, and thus, alleviated the SNL induced behavioral pain (
Figure 2)
[51].
All this evidence supports that melatonin can modulate the triad of chronic pain, sleep disturbance and inflammation. However, detailed clinical studies assessing the impact of Melatonin on levels of various inflammatory mediators in chronic back pain patients with disrupted sleep are lacking.
 +1 point
+1 point