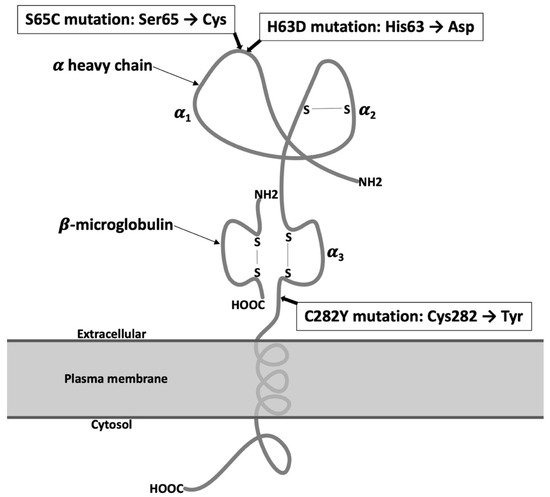
3.1. Regulation of Iron Metabolism in Cardiomyocytes
Our current iron metabolism knowledge and regulation in cardiomyocytes derives from studies on mice with targeted cardiac-specific disruption of iron-related genes
[39][40][41][42]. Under physiological conditions, diferric Tf that binds to the high-affinity TfR1 is used by cardiomyocytes as the main source of iron taken from the serum
[43]. Mouse cardiomyocytes with inactivated
TfR1 gene become severely iron deficient, a phenomenon associated with heart failure and a strongly shortened lifespan
[39]. These abnormalities could be partially reversed by aggressive iron therapy leading to the appearance of non-Tf bound iron (NTBI) and its uptake by cardiomyocytes. Indeed, NTBI is thought to play a major role as a source of iron for cells in various iron overload conditions. NTBI is a multicomponent pool including a considerable proportion of protein-bound iron
[31]. Multiple routes of iron entry into cardiomyocytes exist under iron overload conditions. These include both L-type (LTCC) and T-type (TTCC) calcium channels
[44], proteins from ZIP family and divalent metal transporter 1 (DMT1)
[45]. The entry of iron via all these transporters seems to be beyond the control of canonical regulatory systems of cellular iron homeostasis and may potentially contribute to iron overload in the heart. There is substantial evidence that intracellular iron content can also be regulated through the Fpn-dependent pathway of iron release from cells to the extracellular environment
[46]. Studies on mice with a cardiomyocyte-specific deletion of the
Fpn gene showed a high increase in both iron and ferritin levels in cardiomyocytes
[40]. This cardiomyocytic iron overload was associated with abnormal heart morphology, dilated cardiomyopathy, and reduced survival of
Fpn knock-out mice
[40]. Apart from being influenced by iron fluxes from and to cells, the LIP level is highly dependent on ferritin (Ft), an iron storage molecule with the ability to sequester up to 4500 iron atoms per molecule and thus playing the dual functions of iron detoxification and iron reserve. Ft forms a complex of 24 subunits consisting of a mixture of Ft heavy (H-Ft) and light (L-Ft) chains, showing different functional activities. Heart tissue contains primarily H-Ft-rich ferritins, which contrast to L-Ft-rich ferritins with relatively low iron content
[47]. Importantly, H-Ft-rich ferritins turnover more rapidly and take and release iron more rapidly than L-Ft-rich ferritins
[48]. This may be due to the process of ferritinophagy (autophagic degradation of ferritin), which is mediated by selective cargo-receptor Nuclear Receptor Coactivator-4 (NCOA4) that binds specifically H-Ft subunit and targets the whole protein to emerging autophagosome
[49][50]. Considering accelerated catabolism of heart Ft, it is tempting to suggest that, especially under conditions of sustained and increased iron delivery, the potential of detoxifying iron is much less than in tissues with the main storage function containing up to 90% L-Ft subunits. This may be a reason for the largely reported susceptibility of cardiomyocytes to oxidative stress
[51]. In this context, it tempting to note that the evidence of the manifestation of ferroptosis, a newly identified, dependent on iron and reactive oxygen species form of regulated cell death in iron overload cardiomyocytes, is emerging
[52].
A cardiomyocyte-targeted deletion of
Irp1 and
Irp2 genes in mice resulted in a concerted up-regulation of Fpn and Ft and down-regulation of TfR1 in the heart. They led to the development of cardiac iron deficiency, associated with impaired mitochondrial respiration and cardiac energetics
[42]. Interestingly, IRP-targeted mice were rescued by intravenous iron supplementation.
It is largely accepted that intracellular iron balance also depends on systemic regulation that relies on the interaction of hormone Hepc with Fpn
[53]. The iron regulatory hormone Hepc, synthesized mainly in hepatocytes, limits iron fluxes to the bloodstream by promoting degradation of ferroportin Fpn in target cells, primarily macrophages, enterocytes, and hepatocytes. Thereby, Hepc decreases iron transfer into blood plasma from the duodenum, from macrophages involved in recycling senescent erythrocytes, and iron-storing hepatocytes. However, Hepc derived from hepatocytes may also induce iron sequestration within other Fpn-expressing cells, including cardiomyocytes, which are not essential for maintaining systemic iron homeostasis. Hepc expression has also been reported in many other mammalian cells, including cardiomyocytes
[54]. For the first time, the biological role of tissue-specific Hepc has been demonstrated in mice with cardiomyocyte-specific deletion of the
Hamp gene showing its crucial importance for the regulation of iron metabolism in the heart
[41]. These mice showed cardiomyocyte iron deficiency (associated with fatal contractile and metabolic dysfunction) due to the upregulation of Fpn in cardiomyocytes caused by the absence of negative regulation via cardiac Hepc. Interestingly, circulating liver-derived Hepc did not compensate for the lack of peptide produced locally in the heart
[41].
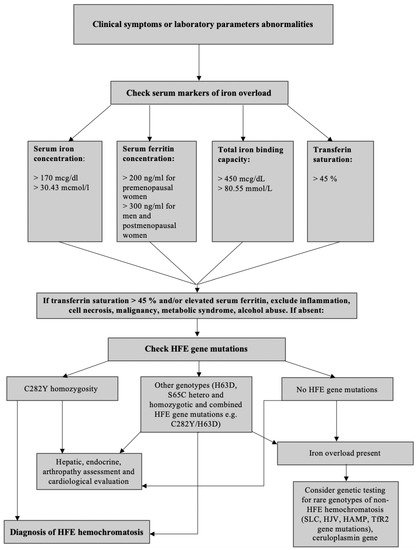
It is known that systemic Hepc insufficiency causes hyperabsorption of dietary iron, hyperferremia, and tissue iron overload, which are hallmarks of HH
[33]. Impairment of the physiological mechanism of regulation leads to Hepc deficiency and, hence, inadequate with the need for increased iron absorption. In this way, primary hemochromatosis differs from secondary iron overload, which develops due to numerous blood transfusions in anemia, e.g., beta-thalassemia, spherocytosis, sickle cell anemia, myelodysplastic syndrome.
The heart is mentioned among organs most charged with iron in various animal models of hemochromatosis
[55]. Precise analysis of iron distribution in cardiac tissue of mice with systemic deletion of
Hamp gene (Hamp
-/-) revealed unexpectedly that iron accumulates mainly in non-cardiomyocytic cells
[40]. Although cardiac iron loading was much more significant in the heart of Hamp
-/- mice than in animals with cardiomyocyte-specific deletion of
Fpn gene, only slightly elevated iron content was detected in cardiomyocytes of Hamp
-/- animals. This finding once again underlies the role of Fpn as a critical regulator of iron content in cardiomyocytes. A summary of the regulation of iron transport in cardiomyocytes is shown in
Figure 2.
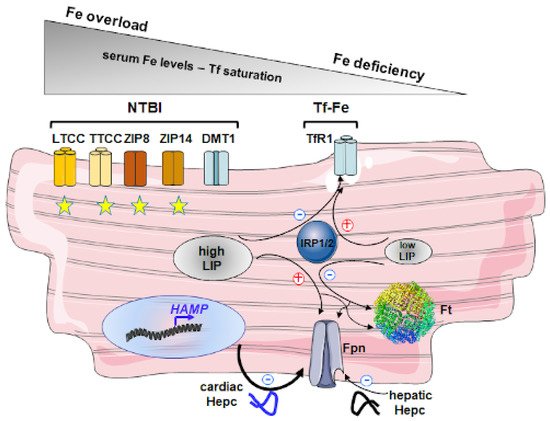
Figure 2. Regulation of iron import and export in cardiomyocytes under iron overload and iron-deficient conditions. In normal and iron-deficient conditions cardiomyocytes acquire iron via transferrin receptor 1 (TfR1)-mediated endocytosis of transferrin-bound iron (Tf-Fe), which is the main source of iron in the blood plasma. At a low iron concentration in the labile iron pool (LIP), direct interactions between IRPs and several IRE motifs stabilize TfR1 mRNA. The converse regulation of TfR1 synthesis, being a consequence of the lack of binding of IRPs to IRE, occurs in cells with high iron in LIP. Iron systemic overload is characterized by both high plasma iron concentration and transferrin saturation and the appearance of non-transferrin bound iron (NTBI). Under these conditions, new routes of iron transport into cardiomyocytes are an option including both L-type (LTCC) and T-type (TTCC) calcium channels, proteins from ZIP family such as ZIP8 and ZIP14. The expression of these importers remains beyond the control by IRP/IRE posttranscriptional mechanism and thus they may potentially contribute to heart overload. Regarding DMT1, only the iron-responsive element (IRE)-containing form, which corresponds to one of two splice forms of DMT1, is responsive to intracellular iron level iron via IRPs and should be down-regulated when LIP is elevated. However, it is not clear whether this form is abundant in cardiomyocytes. Iron in excess of metabolic needs is stored in a soluble and non-toxic form inside ferritin (Ft). In iron-deficient cardiomyocytes, the binding of IRPs to the unique IRE in the 5′-UTR of Ft subunits mRNAs blocks the translation initiation. The converse regulation of Ft occurs in iron-replete cells. Iron export from cardiomyocytes largely depends on ferroportin (Fpn), sole identified cell iron exporter. Its expression in response to iron is regulated intra- and extracellularly. Intracellular regulation involving the IRP/IRE system is the same as that of Ft. Extracellular regulation is mediated by hepcidin (Hepc), a 25-aa peptide predominantly produced by the hepatocytes in response to systemic iron availability. Hepc is also found in heart cells. However, in contrast to hepatic Hepc, iron-dependent mechanisms of the regulation of cardiac peptide are not known. Hepcidin inhibits cellular efflux of iron by binding to Fpn, subsequent internalization, and degradation of Fpn in lysosomes. It has been proposed that retention of iron in cardiomyocytes is mostly due to autocrine regulation by cardiac Hepc, which outcompetes paracrine hepatic Hepc dependent one.
3.2. Molecular Mechanisms of Cardiac Damage in HH
Although many studies have been devoted to the molecular aspect of HH, the mechanism of heart failure induced by iron excess is not known. Overwhelming serum transferrin capacity to bind iron observed in HH patients
[31] leads to noncontrolled iron entry to cardiomyocytes (
Figure 2) and, in turn, may increase cell susceptibility to oxidative stress. Oxidative stress in heart muscle decreases electromechanical coupling, inhibits SERCA2 enzyme function, and leads to the increase in cytoplasmic concentration of Ca ions in cardiomyocytes, leading to impaired relaxation and delayed contraction. Furthermore, oxidative stress induces peroxidation of cellular membranes, including mitochondrial membranes, resulting in a decreased ATP production in oxidative phosphorylation
[56]. Besides, free iron ions may damage mitochondrial and nuclear DNA through the generation of oxidative stress and activates fibroblasts proliferation and differentiation to myofibroblasts responsible for heart fibrosis.
Interestingly, the number of mitochondria in cardiomyocytes is more significant than other cells, hence their greater vulnerability to iron overload, which manifests by the appearance of NTBI in HH patients. From the clinical and practical point of view, it is worth noticing that young adults with HFE-hemochromatosis may experience heart muscle damage when, among laboratory abnormalities, they present mainly high transferrin saturation values without an expected significant increase in the ferritin level. However, the level of NTBI is increased, leading to heart damage at the earliest stages of the disease. Significant hyperferritinemia occurs with the duration of the illness when effective treatment is not initiated.
3.3. Cardiac Involvement in HH
Iron in the heart is mainly located inside the cardiomyocytes rather than in the extracellular matrix, suggesting that iron overload cardiomyopathy is storage rather than an infiltrative process
[1]. The accumulation of iron deposits occurs starting from the epicardial, then through the myocardium to the endocardial layer
[1]. Hypertrophy is one of the macroscopic consequences of the disease. On a mouse model, Sukumaran et al.
[57] analyzed the mechanism of cardiac hypertrophy in HH. It consists of an increase in the expression of heavy myosin chains, with a shift in the number of chains α (decrease) and β (significant increase). The β-chains are responsible for the slower contraction of cardiomyocytes and are a marker of hypertrophy and, as a consequence, diastolic dysfunction. Dilated cardiomyopathy and heart failure are other cardiac complications of the HH, which are explained by different mechanisms. According to the first theory, the lack of
HFE gene function directly impacts myocardial fibrosis
[57]. The increased risk of coronary artery disease due to an increased iron level is the next cause
[58]. According to another hypothesis, the
HFE gene mutation (and the loss of
HFE gene function) could directly affect the appearance of dilated cardiomyopathy
[59]. The last hypothesis assumes the possibility of autoimmune process participation as a cause of heart failure in HH
[60]. All this results in initial hypertrophy, diastolic dysfunction, heart dilatation, systolic dysfunction, and arrhythmias as possible clinical presentations of the HH.
 +1 point
+1 point