1000/1000
Hot
Most Recent
 +1 point
+1 point
α-Synuclein (α-Syn) is a small cytosolic protein associated with a range of cellular compartments, including synaptic vesicles, the nucleus, mitochondria, endoplasmic reticulum, Golgi apparatus, and lysosomes. In addition to its physiological role in regulating presynaptic function, the protein plays a central role in both sporadic and familial Parkinson’s disease (PD) via a gain-of-function mechanism. Because of this, several recent strategies propose to decrease α-Syn levels in PD patients.
The C-terminus is comprised of an EF-hand-like sequence that is negatively charged and is typically unstructured [1]. It is capable of binding Ca 2+ and oxidized dopamine (DA) that affect both normal and pathological properties of the protein. The binding of oxidized DA to α-Syn is non-covalent, reversible, and occurs at the Y125EMPS129 pentapeptide of the C-terminal region with an additional long-range electrostatic interaction with E83 in the NAC region [2][3]. DA-modified α-Syn is less likely to fibrilize and instead forms soluble oligomers that are cytotoxic [4][5]. Ca 2+ binding increases the ability of α-Syn to bind synaptic vesicle membranes [6], at the same time affecting protein ligand binding and oligomerization [7]. Interestingly, overexpression of truncated α-Syn that lacks the C-terminus is sufficient to elicit a PD-like phenotype in mice [8], suggesting that it modulates rather than drives α-Syn-mediated toxicity. Finally, C-terminus harbors a phosphorylation site at serine 129 (pS129). While the physiological role of this post-translational modification is currently unknown, its accumulation under pathological conditions suggests that it may play a role in α-Syn oligomerization or toxicity [9].
The α-Syn protein plays critical roles in both sporadic and familial PD as (1) mutations or multiplications of the SNCA gene cause autosomal-dominant PD [10], (2) genome-wide association studies show a correlation between single-nucleotide polymorphisms in the SNCA locus and the risk of developing sporadic PD [11][12], (3) levels of phosphorylated α-Syn are increased in post-mortem brains of PD patients and patient-derived dopaminergic neurons [13], (4) DA neurons that lack α-Syn are protected in neurotoxin and genetic models of PD [14][15][16]. Additionally, α-Syn has been shown to interact with several proteins implicated in PD and other neurodegenerative disorders, including tau [17][18], LRKK2 [19], Fyn [20], parkin [21], and DJ-1 [22].
Due to the significance of α-Syn in PD pathology, recent advances in therapeutic approaches intended to reduce patient α-Syn levels have gained momentum, including the use of antibodies, vaccines, antisense-oligonucleotides, and small molecules [23][24]. Preclinical studies in cell cultures and animal models have demonstrated successful SNCA gene silencing using antisense oligonucleotides (ASOs), ribozymes, siRNAs, or miRNA without neurotoxic effects [25][26][27][28][29][30][31][32]. Furthermore, several small molecules aimed at preventing α-Syn aggregation (developed by UCB, Proclara and Sun pharmaceutical companies) as well as passive and active immunotherapies that target monomeric or aggregated α-Syn (Prothena, Biogen, AstraZeneca, Takeda, Affiris, Endobody) are currently in clinical trials ( clinicaltrials.gov ).
With the possible exception of immunotherapies that target pathological oligomeric and misfolded forms of α-Syn, such as cinpanemab [33], prasinezumab [34], and ABBV-0805 (currently in phase I clinical trial), these approaches are expected to deplete monomeric α-Syn. As this protein is abundantly expressed in the brain and the periphery, changes in normal function and potential side effects of decreased α-Syn levels need careful evaluation. For example, multiple studies demonstrate roles for α-Syn in regulating dopamine neurotransmission both ex vivo (reviewed in [35]) and in anesthetized animals in vivo [36][37], indicating that α-Syn deficiency might lead to synaptic dysfunction. Although α-Syn knockout mice (α-Syn −/− ) are viable and fertile, these animals exhibit decreased behavioral responses to amphetamine [38] and reduced learning ability in tests requiring both working and spatial memory [39]. Moreover, despite the evidence for neuroprotection in α-Syn −/− mice, acute α-Syn knock-down in adult rats unexpectedly resulted in neuroinflammation and degeneration of nigral dopaminergic neurons [40][41][42]. It may be that the developmental stage in which α-Syn depletion occurs is important, or additional mechanisms govern protective or deleterious consequences of protein expression. In either case, these contrasting reports highlight the importance of understanding the physiological role of the protein and the consequences of its decreased expression, particularly in mature animals.
Such promiscuity of α-Syn localization and interactions makes it difficult to isolate a single mechanism of action. While cellular organelles and pathways involved in α-Syn physiological function are highlighted in several recent reviews [35][43], here we focus on an aspect of α-Syn normal function that has been relatively ignored—the role of the protein in transcriptional control and immediate early genes induction in neurons. Several lines of indirect evidence support the pathophysiological relevance of α-Syn in transcription. Microarray analysis of gene expression in neuroblastoma cell lines transfected with WT α-Syn revealed 15 genes that were upregulated and 30 that were downregulated [44]. Similarly, RNA-seq analysis of induced pluripotent stem cells (iPSC)- derived dopaminergic neurons from PD patients carrying a triplication of the SNCA locus identified 131 upregulated and 115 downregulated genes compared to neurons from healthy control [45]. While, unsurprisingly, some of these changes can be attributed to stress response and other processes downstream of α-Syn-induced aggregation and toxicity, there is evidence supporting a role for the protein in directly affecting gene expression as discussed below.
In contrast to direct binding to polynucleotides, α-Syn may affect gene expression by altering DNA methylation. Post-translational modification of DNA and histones are key mechanisms of epigenetic control of gene expression that in addition to playing a role in synaptic plasticity and development are involved in multiple neurodegenerative disorders [46][47]. Typically, methylation of CpG-rich promoter regions by DNA methyl transferases (DNMT) represses gene transcription, which is countered by passive DNA demethylation that restores promoter activity [48]. Association of α-Syn with DNMT1, an enzyme that is normally located in the nuclei, causes its extrusion to the cytosol, resulting in global DNA hypomethylation ( Figure 1 ). In human and mouse brain, this was shown to affect the expression of multiple genes including SNCA , SEPW1 , and PRKAR2A [49].
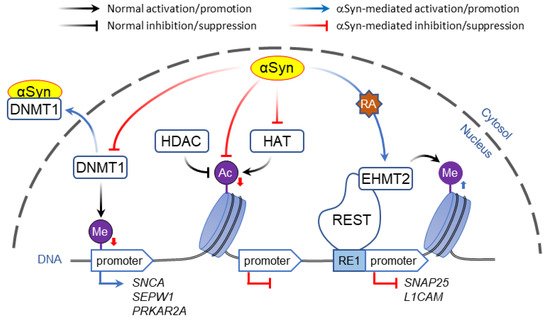
Whereas histone acetylation is thought to always promote gene expression, histone methylation regulates transcription depending on the context. For example, H3 methylation of lysine at position 4 (H3K4 modification) activates the nearby DNA loci, whereas modifications of the same histone at positions 9 or 27 (H3K9 and H3K27) inactivate gene expression.
Activity-dependent opening of voltage-gated calcium channels provides another mechanism that allows to encode acute information flow into changes in gene expression that form the basis of neural plasticity. In particular, L-type calcium channels (LTCC) are uniquely positioned to couple neuronal activity and immediate early genes expression ( Figure 2 ) [50][51].
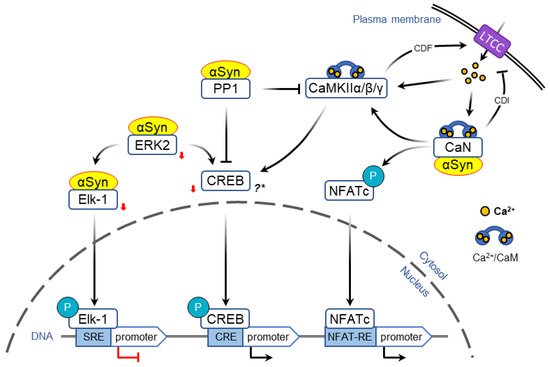
Although multiple studies implicate an involvement of α-Syn in transcription, it is unclear whether these pathways are physiologically relevant in humans. An important distinction needs to be drawn between studies of α-Syn overexpression/mutation, which are meant to emulate synucleinopathies, and those where the protein is depleted, which are intended to address its normal function. While several cellular regulators of transcription have been shown to mediate toxicity induced by α-Syn overexpression, it is unknown if decreasing α-Syn levels will have an opposite effect on gene expression. Similarly, even though reports of deficits in working and spatial memory in α-Syn null mice support a role of the protein in learning [39], further studies are required to determine the effects of therapeutic doses of anti-synuclein medications and more specific tests for the immediate early genes expression. Finally, while it is abundantly expressed in the brain, α-Syn is also expressed in the gut, liver, lungs and is necessary for the normal development and functioning of these tissues [52]. Silencing α-Syn in the periphery may cause side-effects by affecting the expression of genes in non-neuronal cells, a possibility that should be investigated in future studies.





