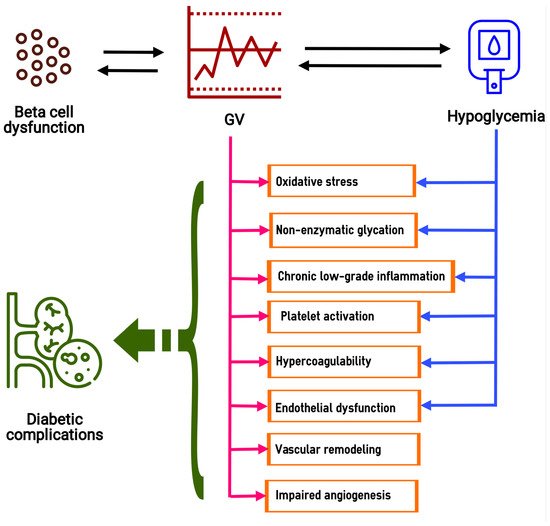
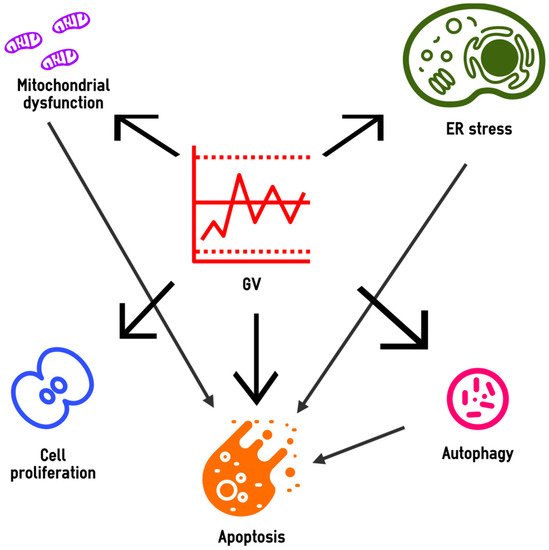
4.1. Gene Expression
Although there are scarce data on changes in gene expression induced by excessive GV, there is a large pool of studies on gene profiling related to hyperglycemia. Using high-throughput technologies, differential gene expression was measured under hyperglycemic conditions in beta cells
[140][141], pancreatic cells
[142], hepatic cells
[143][144], endothelial cells
[145], myotubes
[146], cardiomyocytes
[147], vascular smooth muscle cells
[148][149], adipose progenitor cells
[150], kidney cells
[151], renal tubular epithelial cells
[152], retina
[153][154], immune cells
[155][156] and others. The genes that demonstrate an altered expression in hyperglycemia are mostly involved in glucose metabolism, inflammation and immune processes, endothelial dysfunction, angiogenesis, oxidative stress, mitochondrial dysfunction, hypoxia and cell death.
Transcriptomic studies have revealed the effect of hyperglycemia on the expression profile of a large number of genes. More than 80 genes involved in hepatic lipid metabolism were differentially expressed in hyperglycemic rats with a model of T1D
[143]. With the use of high-throughput RNA sequencing, it was demonstrated that hyperglycemia has a strong effect on HepG2 cells, with 4259 genes showing a differential expression. These genes participate in cholesterol metabolism, DNA replication, complement and clotting cascades
[144]. Maier et al. hypothesized that hyperglycemia amplified the expression of genes induced by thrombospondin-1 in vascular smooth muscle cells. Microarray analysis revealed that hyperglycemia altered the expression of 30 genes, while hyperglycemia combined with thrombospondin-1 altered the expression of 2822 genes. These findings suggest that hyperglycemia may significantly enhance the thrombospondin-1 effect on atherosclerosis progression
[148].
Fewer studies have focused on gene expression in hypoglycemia. In sirtuin 6-deficient mice that developed a lethal early-life hypoglycemia, the microarray revealed nearly 200 genes with an altered expression. These genes were involved in glucose metabolism, nutrient stress responses, glycolysis and mitochondrial function
[157]. A gene response to insulin-induced hypoglycemia was estimated in the mouse retina by an array. Genes whose expression was modified by low glucose were enriched in lysosomal function, glutathione metabolism and apoptotic pathways and potentially involved in retinal cell death
[158]. A set of genes specifically activated by recurrent hypoglycemia was revealed in a study of whole genome expression profiling after recurrent hypoglycemia and acute hypoglycemia in the adrenal medulla of normal Sprague Dawley rats. These genes were related to the activation of the unfolded protein response, impaired epinephrine secretion, increased neuropeptide signaling, altered ion homeostasis and down-regulation of genes involved in Ca
2+-dependent exocytosis of secretory vesicles
[159].
It was found that even short-term enhanced GV could adversely affect gene expression in the arterial wall. In the study of Zervou et al., pIns-c-MycER(TaM) transgenic mice were successively exposed to hypo- and hyperglycemia, after which they recovered for up to 3 months. The expression of 95 genes was significantly affected by hypoglycemia, and 769 genes were affected by hyperglycemia. These genes were involved in atherogenic processes, including inflammation and arterial calcification. Although the expression of many genes returned to its initial level after 3 months, in one in four genes, recovery was not observed
[160]. These data indicate that non-physiological glucose fluctuations may have a prolonged effect on gene expression. Further research in this direction is urgently needed.
Recently, we performed the bioinformatic reconstruction and analysis of the gene network of GV. The network consisted of 37 genes/proteins associated with both hyperglycemia and hypoglycemia. GV-related molecules were involved in glucose metabolism, intracellular signaling, cell proliferation and other biochemical/physiological processes; they were identified in the central positions of the gene networks of diabetic vascular complications
[161].
4.2. Epigenetic Modifications
Glucose can induce a number of epigenetic modifications that significantly alter the functioning and vital activity of various cell types. In a pivotal work in this field, El-Osta et al. demonstrated that transient elevation in the glucose level causes long-lasting epigenetic changes in the NF-κB subunit p65 promoter in aortic endothelial cells in vitro and in non-diabetic mice. These changes were associated with an increased p65 gene expression that persisted for at least 6 days of subsequent normal glucose levels, and NF-κB-induced increases in MCP-1 and VCAM-1 expression
[162]. These data clearly indicate that epigenetic modifications may be an important mechanism in GV-induced vascular inflammation and dysfunction.
Costantino S. et al. found DNA hypomethylation and histone 3 acetylation on the p66
Shc promoter of the SHC-transforming protein 1 gene
(SHC1), resulting in gene overexpression, in patients with T2D. CGM-derived MAGE and postprandial glucose, but not HbA1c, were associated with the epigenetic profile. The intensification of glycemic control over 6 months did not eliminate the changes
[163]. The mechanism of p66
Shc-reduced CpG methylation could be related to methyltransferase DNA (cytosine-5-)-methyltransferase 3 beta (DNMT3b), an enzyme playing an important role in the maintenance of DNA methylation. Sirtuin 1 (SIRT1) could be involved in H3 deacetylation of p66
Shc[163][164]. In patients with T2D, the expression of DNMT3b and SIRT1 was inhibited compared to the control
[163].
Recently, the effect of glucose on whole genome DNA methylation was studied in human retinal endothelial cells and HUVECs
[165]. The lines were exposed to basal (5 mmol/L) or high (25 mmol/L) glucose-containing media for variable lengths of time. When comparing the endothelial cells, incubated for 2 days versus 7 days, 17,354 and 128 differentially methylated CpGs in 88 and 8 differentially methylated regions were identified for HUVECs and retinal endothelial cells, respectively. Pathway enrichment analyses highlighted the involvement of regulators of embryonic development (i.e., HOX genes), TGF-β signaling, bone morphogenetic protein (BMP) signaling, runt-related transcription factor 2 (RUNX2) transcriptional regulation and the complement cascade.
It was demonstrated that fluctuating glucose significantly decreased the phosphorylation of the endothelial nitric oxide synthase (eNOS) at Ser-1177 and increased the phosphorylation of JNK and p38, leading to the damage of vascular endothelial cells
[166]. IHG lowered the phosphorylation levels of protein kinase B (v-akt murine thymoma viral oncogene homologue, Akt), AMPK and glycogen synthase kinase 3 beta (GSK3β), influencing the function of endothelial cells and beta cells
[19][91][167].
Small single-stranded non-coding RNAs (miRNAs) have been discussed as another method of epigenetic regulation. Aberrant miRNA expression is implicated in the pathogenesis of numerous diseases, including diabetes and its complications
[168]. In HUVECs cultured under IHG conditions, 13 miRNAs were differentially expressed. miR-1273g-3p partially mediated the effect of IHG on the autophagy, migration and proliferation of HUVECs
[74]. Another example of GV-induced miRNA-dependent changes is a phenotype polarization switch of microglia. In microglial cells, glucose fluctuations induce polarization transitions from M2 to M1. The M1 phenotype has proinflammatory effects and can be responsible for neuronal damage; in contrast, M2-polarized microglia can inhibit the inflammatory response and promote nerve repair. It was found that miR-124, miR-145, miR-146a and miR-711 are implicated in the M2 phenotype polarization of microglia, while miR-689 and miR-155 are involved in M1 polarization. In macrophages, miR-124 and miR-146a induced M2 phenotype polarization
[169]. In the glucose fluctuation cell model, miR-129-3p suppressed glucose-mediated hippocampal neuronal damage. Specifically, miR-129-3p overexpression produced a dramatic reduction in calcium overload, ROS generation and an increase in antioxidant activity
[170]. In cultured HUVECs, miR-1273g-3p mediates the effect of GV on autophagy and endothelial dysfunction
[74]. In human endothelial cells, miR-185 and miR-21 were induced by oscillating glucose, leading to an impaired antioxidant response by the dysregulation of glutathione peroxidase 1 and superoxide dismutase 2
[97][171]. It was demonstrated that IHG induces the up-regulation of HIF-1α and miR-210 in glomerular mesangial cells, which might play a pivotal role in the series of molecular events triggered by GV
[22].
Thus, the effects of glucose fluctuations on gene expression can be exacerbated and prolonged by epigenetic modifications. At present, glucose-induced epigenetic modifications and related changes in the activity of signaling pathways are considered as an important mechanism of “metabolic memory” or “metabolic karma” in diabetes
[168][172][173].
4.3. Signaling Pathways
The cellular and molecular effects of GV are realized through a variety of signaling pathways. The activation of PKC is among the initial molecular events under high-glucose conditions. PKC is a driver of numerous signal transduction cascades that regulate cell metabolism and plasticity. Among the downstream targets of PKC is NADPH oxidase that activates superoxide production and thus exacerbates oxidative damage
[174].
A number of molecular effects of oxidative stress are mediated via NF-κB-dependent signaling pathways. NF-κB is a universal transcription factor that controls the expression of genes for the immune response, apoptosis and cell cycle. In diabetes, ROS, AGEs and angiotensin II induce an inflammatory response, endothelial dysfunction and renal fibrosis via the activation of NF-κB. Accordingly, NF-κB is considered as a potential target in diabetic vascular complications
[175]. As it has previously been mentioned, transient high glucose induces prolonged NF-κB activation in vascular endothelial cells
[162]. The IHG-stimulated activation of NF-κB in cultured HUVECs down-regulated the expression of Bcl-2, an antiapoptotic protein
[176]. In vascular cells, glucose fluctuations promote the dysfunction of large-conductance, calcium-activated potassium channels via the overproduction of ROS and activation of PKCα/NF-κB/MuRF1 signaling
[177]. ROS-mediated NF-κB activation under high-GV conditions up-regulates the receptor for AGEs in podocytes
[22].
The dysregulation of the phosphoinositide-3-kinase (PI3K)/Akt, mitogen-activated protein kinase (MAPK) and AMPK pathways is considered to be involved in altered glucose metabolism and related biochemical abnormalities in diabetes and high GV
[178][179]. The PI3K/Akt signaling pathway, which is essential for cell survival and growth, plays an important role in preventing endothelial cell injury induced by high glucose. It was shown that IHG induces a more severe decrease in the phosphorylation of Akt and GSK-3β than CHG in cultured HUVECs; this effect is associated with reduced cell viability
[19]. In agreement with these data, it was demonstrated that IHG suppresses NO synthesis in cultured HUVECs to a greater extent than CHG via the inhibition of PI3K/Akt and eNOS activity
[58]. The pro-apoptotic effect of IHG in cultured neuronal cells (PC12 cell line) also involves the PI3K/Akt pathway
[133]. The oxidative and inflammatory stress and microglial activation induced by acute glucose fluctuation in the mouse microglial BV-2 cell line were mediated through the PI3K/Akt, NF-κB and MAPK cascades
[134].
MAPK families play an important role in cell proliferation, differentiation and apoptosis. The MAPK families include extracellular signal-regulated kinase (ERK), JNK and p38 MAPK
[180]. Some data point to the role of these signaling molecules in the realization of the effects of GV in the target organs. It was demonstrated that MAPK (ERK1/2), as well as the PI3K and NF-κB signaling pathways, is involved in the proliferative effect of IHG in VSMCs
[138].
In vascular endothelial cells, IHG increased the phosphorylation of JNK
[166]. The JNK pathway plays a central role in the cell response to hyperglycemia, oxidative stress, proinflammatory cytokines and other stress-inducing stimuli. The JNK-dependent effects include the regulation of gene expression, cell death and cellular senescence
[181][182]. In patients with diabetes, JNK contributes to vascular insulin resistance and endothelial dysfunction
[183]. It was demonstrated both in vivo and in vitro that the PKC/JNK pathway mediates the pro-apoptotic effect of glucose fluctuations in endothelial cells
[129][184].
In diabetes, high glucose activates p38 MAPK signaling
[181]; high GV may be an additional trigger of the event
[166]. It was shown that GV generates the more severe up-regulation of type I collagen synthesis and fibrosis of aorta via the activation of the ROS/p38 MAPK/Runx2 pathway in Sprague Dawley rats with streptozotocin-induced diabetes
[185]. In astroglial cells, glucose fluctuations induce toxicity with oxidative and inflammatory stress by activating p38 MAPK and NF-κB
[99].
The interactions among the MAPK, NF-κB and TGF-β/Smad signaling pathways are essential for fibrogenesis. It is well known that TGF-β’s biological effects were realized by activating downstream mediators Smad2 and Smad3, which is negatively regulated by an inhibitory Smad7
[186]. The activation of the MAPK/ERK and TGF-β/Smad signaling pathways is considered as a cornerstone in the pathogenesis of renal fibrosis in diabetic kidney disease
[187]. As it was demonstrated in mice with alloxan diabetes, excessive blood glucose fluctuations cause the more pronounced activation of the TGF-β/Smad2 and ERK/MAPK pathways in the kidney compared to stable hyperglycemia. These changes in signal transduction were accompanied by the marked increase in type I collagen synthesis and suppression of collagen degradation
[188]. The inhibition of skin collagen synthesis and increase in collagen degradation under high GV is also attributed to both the MAPK and Smad signaling pathways
[189].
AMPK is a master regulator of metabolism which acts as an intracellular sensor of energy availability
[178]. The glucose shortage promotes AMPK activity; meanwhile, overnutrition inhibits it. In many cell types, AMPK stimulates glucose uptake via trafficking glucose transporters GLUT1 and GLUT4; acutely stimulates glycolysis; and, in the longer term, tends to promote oxidative metabolism. The activation of autophagic flux via ULK1 is considered as an important AMPK-dependent mechanism of cellular metabolic adaptation
[190][191]. Recently, it has been demonstrated that high glucose represses AMPK signaling via MG53 E3 ubiquitin ligase-mediated AMPKα degradation and deactivation
[192]. Currently, little is known about the effect of GV, which is characterized by intermittent glucose excess and deprivation, on AMPK activity in diabetes. It was found that the activation of AMPK by globular adiponectin can inhibit, at least partially, the IHG-induced apoptosis of HUVECs
[193].
mTORC1 is another protein kinase that is regulated by glucose availability; however, unlike AMPK, mTORC1 is activated in high-glucose conditions. When it is activated, mTORC1 shifts the metabolic paradigm towards anabolic processes, promotes cell growth and suppresses autophagic flux
[191]. It was demonstrated that the inhibition of AMPK by high glucose inversely correlates with the activation of the mechanistic target of rapamycin (mTOR) pathway in beta cells
[194]. It is currently known that upon glucose depletion, mTORC1 is inhibited by AMPK-dependent and AMPK-independent mechanisms
[195]. Recent data indicate that aldolase could be a sensor for both low and high glucose levels, linking to the AMPK and mTORC1 pathways
[196]. In cancer, diabetes and other diseases characterized by abnormal glucose metabolism, mTORC1 is deregulated
[195][197]. In diabetes, hyperactivated mTORC1 is involved in the pathogenesis of cardiomyopathy
[198], diabetic retinopathy
[199] and diabetic kidney disease
[200]. Unfortunately, the role of mTORC1 signaling in GV-related vascular effects has not been studied to date.
Thus, the deteriorative effects of high GV in the target cells are realized through the PI3K/Akt, NF-κB, MAPK (ERK), JNK, TGF-β/Smad and other signaling pathways (Table 1). Elucidating the pathophysiological role of AMPK and mTORC1 under fluctuating glucose conditions is a promising challenge for future research.
Table 1. The principal signaling pathways mediating the pathophysiological effects of high GV in diabetic complications.
| Effect |
Pathways |
Refs. |
| Oxidative stress in endothelial and neural cells |
PKC/NF-κB, PI3K/Akt, p38MAPK |
[99][134][174][175] |
| Endothelial dysfunction and apoptosis |
PI3K/Akt, NF-κB, PKC/JNK |
[19][58][129][176][184] |
| Proliferation of VSMCs |
MAPK (ERK1/2), PI3K/Akt, NF-κB |
[138] |
| Vascular low-grade inflammation |
NF-κB and p38 MAPK |
[162] |
| Renal fibrosis |
MAPK (ERK1/2) and TGF- β/Smad |
[188] |
| Aortic fibrosis |
TGF-β/Smad, NF-κB, p38 MAPK and Runx2 |
[185] |
| Neuronal apoptosis and neurodegeneration |
PI3K/Akt, NF-κB |
[133][134] |
 +1 point
+1 point