Inherent to its secretory phenotype, the placenta is also a target of hormonal biological activity because it expresses most receptors for these hormones and growth factors. Therefore, placental hormones act in endocrine, paracrine, and autocrine pathways in the Maternal-placental-fetal-unit (MPFU).
2.1. The Insulin and IGF-I/IGF-II Axis and Molecular Pathways Primarily Disturbed in GDM Placentae
Insulin resistance is one of the first alterations in the pathogenesis of GDM. Insulin acts through two known receptor isoforms: insulin receptor A (IR-A) and insulin receptor B (IR-B). These are heterodimer receptors composed of an extracellular α-subunit that binds to insulin, and an intracellular β-subunit that binds to the insulin receptor substrate 1 (IRS-1). Both isoforms are transcripts of the same
INS gene. However, the IR-A product lacks a sequence of 36 nucleotides in the C-terminal of the α-subunit as a result of alternative splicing of exon 11. In contrast, IR-B is the result of the full transcription of the
INS gene
[36][37]; this explains the differences in their activities. Insulin has a higher affinity for IR-A than for IR-B. In addition, IR-A activates the Ras-Raf-mitogen-activated protein kinase (MAPK) cascade, whereas IR-B activates the phosphatidylinositol 3-kinase (PI3K) and protein kinase B (Akt) signaling.
The study of the cellular pathways activated by insulin began around 33 years ago
[38]. Since then, diverse research papers have scrutinized these cellular cascades, and finally, in 1995, the role of PI3K and Akt in the glycemic control and other metabolic activities of insulin was described
[39][40]. After IR-B binding to insulin, PI3K binds to tyrosine-phosphorylated IRS proteins, leading to the formation of phosphatidylinositol (3,4,5)-triphosphate (PIP3). Downstream effects of PIP3 lead to activation of 3-phosphoinositide dependent protein kinase (PDK)1 and the subsequent activation of a variety of kinases, of which Akt 1–3 is the best-studied
[41]. Main metabolic activities of insulin are related to Akt phosphorylation: (i) increased glycogen synthesis by inactivation of glycogen synthase kinase-3 (GSK3) α/β and activation of glycogen synthase
[39]; (ii) Decreased transcription of gluconeogenic genes in liver and autophagy genes in muscle, by phosphorylation of forkhead box (FOX) transcription factors
[42]; (iii) stimulated protein synthesis and suppression of autophagy by phosphorylation of tuberous sclerosis 2 (TSC2) and the 40 kDa proline-rich Akt substrate (PRAS40) which leads to activation of mTORC1
[43][44]; (iv) increased glucose uptake by phosphorylation of TBC1 domain family member 1/Akt substrate of 160 kDa (TBC1D4/AS160) which regulates trafficking and translocation of GLUT4 cytoplasmic vesicles to the plasma membrane
[45]. In line with their metabolic effects, IR-B is preferentially expressed by classical insulin-sensitive cells such as hepatocytes, adipocytes, and myocytes, which have important roles in glucose, lipid, and protein metabolism.
On the other hand, activation of IR-A induces the Grb-2/Erk 1/2 MAPK pathway related to cell growth, differentiation, and survival processes
[46]. This isoform is predominantly expressed in cancer tissues, the brain, hematopoietic cells, and the placenta
[36].
Although the human placenta expresses both insulin receptors, IR-A is expressed at higher levels than IR-B
[47]. This differential ratio is probably related to the need for tight control of the crucial proliferative and pro-differentiation pathways during pregnancy. On the other hand, there are redundant placental pathways to help in the vital fetoplacental glucose transfer, besides insulin/IR-B mediated GLUT-4 translocation
[48][49], as occurs in insulin-dependent tissues. During the first trimester of pregnancy, IR-A is expressed more in the apical membrane of syncytiotrophoblasts whereas at term IR-B is concentrated in endothelial cells of the villi microvasculature
[50].
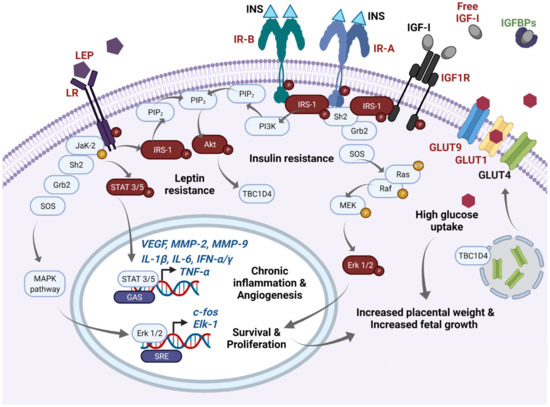
In
Figure 1, we summarize the disturbed molecular pathways in GDM placentae. Immunohistochemical and blotting studies showed that GDM placentae have a lower total protein expression of IR-A, PIP3, and IRS-1 in comparison to placentae from uncomplicated pregnancies. Interestingly this occurs independently of the metabolic control of the disease which implies profound and sustained effects in placenta signaling networks
[51][52]. Also, it seems that the obesogenic environment is an additional regulating factor of insulin signaling among GDM placentae, involving IRS-2, PI3K, and GLUT4, the most sensitive targets to obesity
[53]. Additionally, it has also been reported that GDM placentae have a pronounced phosphorylated pattern of IR and IRS-1 proteins, accompanied by hyperphosphorylation of STAT-3, MAPK 1-3 (Erk 1/2), and Akt
[54][55]. Altogether, these data suggest that GDM primes the placenta to overstimulate the insulin signaling to compensate sustained exposure to hyperglycemia. The deficit in the total protein levels of these mediators results in insufficient placental glucose uptake, and consequently in a hyperglycemic state
[56][57]. Although, other authors did not observe differences in the total protein levels of placental IR and IRS-1
[54][58]. Considering these inconsistencies, we believe more studies are needed to clarify insulin signaling in GDM placentae and to understand how placental imbalance in these signaling pathways results in higher levels of inflammatory cytokines, adipokines and oxidative reactive species, insulin resistance, and vascular disorders, all of which prevail in the local placenta and peripheral tissues of GDM mothers.
Figure 1. Main molecular pathways disturbed in GDM placentae. In this schematic, we highlight in red main molecules reported as overexpressed/overactivated in GDM placentae, whereas downregulation is highlighted in green. High expression of phospho-IR-A and phospho-IR-B has been reported in the GDM placenta. After insulin binding, IR-A phosphorylates IRS-1 and recruits Scheme 2. and Grb2 proteins which induce the GTPase activity of Ras, and then a GDP is exchanged by a GTP. This initiates a subsequent cascade of phosphorylations of Raf, MEK, and Erk 1/2. Finally, overexpressed phospho-Erk proteins translocate to the nucleus and recognize SRE sites favoring active transcription of c-fos and Elk-1. FOS proteins have been implicated as regulators of cell proliferation, survival, differentiation, and transformation. This pathway mediates increased placental weight and increased fetal growth in GDM. Additionally, increased levels of free IGF-I resulting from low IGFBPs serum concentrations also activate IR-A signaling as well as IGF1R and are related to proliferative effects. IGF-I excess has been also implicated in macrosomia and excessive placental growth in GDM women. On the other hand, after insulin binding to IR-B, phospho-IRS-1 activates PI3K and leads the formation of PIP3 from PIP2. Then, PIP3 activates Akt which mediates diverse metabolic effects; one of them includes GLUT4 translocation from endosomes to the cellular membrane through TBC1D4 signaling. High expression of GLUT1 and GLUT9, and probably GLUT4, mediates high glucose uptake in the GDM placenta, which can also participate in fetal and placental growth through an excess of this energetic substrate. Finally, GDM placentae present high expression of leptin and its receptor. High expression of phospho-leptin receptor recruits JaK-2 protein and activates STAT 3 or 5 proteins. Then, STATs dimerize and translocate to the nucleus and recognize GAS sites and provoke transcription of VEGF, MMP-2, MMP-9, TNF-α, IL-1α, IL-1β, IFN-α, IFN-γ, among others, which exacerbate the inflammatory placental milieu and contribute to stimulating the angiogenic process. Additionally, activation of the leptin receptor can also crosstalk with MAPK and Akt pathways. Sustained overactivation of all these pathways finally leads to clinical insulin and leptin resistance. GAS: Gamma-activated sequence. GDM: Gestational diabetes mellitus; GLUTs: Glucose transporters; IGFBPs: IGF-I binding proteins; IGF-I: Insulin-like Growth Factor 1; IGF1R: IGF-I receptor; INS: Insulin; IR-A: Insulin receptor type A; IR-B: Insulin receptor type B; LEP: Leptin; LR: Leptin receptor; SRE: Serum response elements.
Another system that can cross-react with insulin signaling is the IGF-I axis. The system consists of two ligands IGF-I and IGF-II, two receptors IGF-1R and IGF-2R, six IGF binding proteins (IGFBP 1–6), and four insulin-like growth factor binding protein-related peptides IGFBP-rP1-4
[55][59]. These growth factors mainly regulate growth and metabolism throughout the life cycle, but its activity is critical during intrauterine life for mammalian development. They provide a signal to cells to indicate that adequate nutrients are available, and therefore to enhance cellular protein synthesis, to favor hypertrophy, and to stimulate cell division
[60].
Insulin and IGF-I present a relatively low homology of 34% (BLAST alignment: CAA40342.1 and AAN39451.1), but their receptors are highly homologous, around 57% (BLAST alignment: CAA28030.1 and AAA59452.1). These similarities in their structures generate promiscuous interactions between them. IGF-II binds to IR-A with an affinity close to that of insulin, but it does not bind to IR-B
[61]. Additionally, random hybrids of IR-A/IR-B and hybrids of IRs/IGF-1R have been reported in the placenta
[36][62][63]. IGF-1R/IR hybrids bind IGF-I and IGF-II with high affinity but bind insulin with a relatively low affinity
[64].
Given the structural similarities between components of the IGFs/Insulin axis, it is not surprising that these hormones share signaling pathways. IGF-I binds to IGF-1R and activates two main cascades: (i) PI3K/Akt pathway via IRS-1 phosphorylation which predominantly leads to metabolic effects; (ii) Ras-Raf-MAPK pathway via SHC domain proteins which control cellular growth and differentiation
[59]. On the other hand, IGF-II binds with high affinity to IGF-2R, also known as the cation-independent mannose 6 phosphate receptor. This interaction targets IGF-II for its lysosomal degradation and consequently, IGF-2R sequesters IGF-II, controlling the circulating levels of this hormone. Therefore, the biological activity of IGF-II is exclusively derived from its binding to IR-A or IGF-1R, considering that it does not bind to IR-B, as mentioned before
[65].
The placenta synthesizes all components of the IGF axis from early stages at 7 weeks of gestation. IGF-I was more expressed in the second and third trimesters of pregnancy in comparison with early pregnancy, and it was expressed by practically all placental cells except syncytiotrophoblasts. Whereas IGF-II was profusely expressed by cytotrophoblasts, mesoderm core, basal plate, columnar cytotrophoblasts, amnion, and chorion. IGF-IR was expressed ubiquitously in the placenta, except in Hofbauer cells. All six IGFBPs were expressed in decidua basalis and parietalis
[66].
One key insulin-like action of IGF-I is related to glucose metabolism
[67]. Biomedical and clinical studies indicate that IGF-I is a hypoglycemic factor that increases glucose uptake in different kinds of cells, including euglycemic trophoblasts
[68][69][70]. However, deeper studies are needed to confirm its metabolic effects in the GDM milieu. In contrast, IGF-II seems to present a hyperglycemic effect since overexpression of IGF-II in pancreatic β-cells results in the development of T2DM
[71].
Excessive fetal growth and weight is a common complication from GDM newborn babies. Macrosomia has been explained by two central modulators: hyperglycemia and activation of the IGF-I axis. Maternal hyperglycemia increases energetic substrate availability and then stimulates excessive growth and adiposity in GDM mothers
[72]. In fact, an increased concentration of glucose transporters GLUT1 and GLUT9 has been observed in GDM placentae, which favors an increased placental and fetal D-glucose uptake
[49][52][73][74] (see
Figure 1).
Concerning IGF-I signaling, different serum components of the pathway have been measured in GDM patients in the last 20 years. In a recent meta-analysis developed by Dr. Wang’s group, in which they analyzed 12 independent studies, they found GDM was consistently associated with higher maternal IGF-I levels in mid-gestation (20–29 weeks) and late-gestation (>30 weeks), whereas serum IGF-II did not present significant changes between GDM and control mothers
[60]. Interestingly, most data show significantly lower cord levels of IGFBP1, IGFBP2, IGFBP3, IGFBP-6 or IGFBP-rP1
[55][75][76], and lower maternal levels of IGFBP1 and IGFBP2
[55]. All IGFBPs bind both IGF-I and IGF-II with similar affinities (except IGFBP-6 which is essentially IGF-II specific
[77]). Their metabolic effects are related to inhibition of IGF-I signaling by sequestrating it into a circulation reservoir. Consequently, diminished levels of IGFBPs and IGFBP-rPs result in higher cord blood levels of free-IGF-I in GDM patients
[75]. Further, the research group of Dr. Sciacca identified an increased phosphorylation pattern of IGF-1R in placentae from metabolic uncontrolled mothers with GDM and T2DM
[58]. These changes support a persistently activated IGF-I signaling in GDM placentae by increased activity of free-IGF-I (see
Figure 1).
It is well known that the growth hormone (GH)-IGF-I axis is the major regulator of longitudinal growth along life. In addition, significant and positive correlations between the birth weight of newborns from GDM mothers and maternal serum IGF-I or molecules of the IGF-I signaling in GDM placentae have been described
[52][78][79]. Therefore, overactivation of IGF-I signaling may be one critical factor involved in the development of macrosomia in babies from GDM mothers. Other morphologic changes in the placenta are also related to macrosomia, including broader intervillous spaces, increased terminal villus volume, a large proportion of immature villi, and a larger syncytiotrophoblast surface allowing higher amounts of glucose to cross the placenta
[20][21].
One additional hypothesis in excessive fetal weight gain in GDM is related to IR/IGF-1R hybrids. A high proportion of these hybrids has been reported in skeletal muscle and adipose tissue of T2DM patients
[63][80][81], and in placentae from insulin-resistant women
[82]. IR/IGF-1R hybrids increase the binding sites for IGF-I and IGF-II, favoring IGF signaling, proliferation, and anabolic processes, as has been previously published in cancer models
[83]. However, this hypothesis, which deserves to be further explored, has not so far been studied in the placentae of mothers with GDM.
2.2. The Role of Pancreatic β-Cells and Lactotroph Hormones in GDM
In a healthy pregnancy, insulin resistance increases between 50% and 60% in the third trimester, compared to the pre-pregnancy period
[84]. This physiologic resistance is needed to ensure adequate delivery of glucose to a fast-growing fetus. In response, the mother needs to expand her capacity for insulin secretion which is achieved by an increase in β-pancreatic cell mass and number, finally leading to a euglycemic pregnancy. The lactotroph hormones prolactin (PRL) and human placental lactogens participate in cell-specific β-cell responses to counteract the physiological insulin resistance developed during pregnancy. These pancreatic adaptations occur before the onset of insulin resistance in pregnancy
[9].
The maternal decidua is the main extra-pituitary source of PRL synthesis
[85][86], although columnar trophoblasts and villous cytotrophoblasts of the placenta can also synthesize it to a lesser extent
[85][87]. On the other hand, placental lactogens are exclusively synthesized during pregnancy by fetal syncytiotrophoblasts and include human placental lactogen (hPL), human chorionic somatomammotropin A and B (hCS-A and hCS-B), and the placental growth hormone (PGH). Evolutionary studies indicate that placental lactogens are closely related in their chemical structure to the human Growth Hormone (GH) as a result of three duplications and one deletion in the
GH gene
[88]. Derived from this structural homology, lactogens share with GH their binding capacity to both somatogenic and lactogenic receptors
[89]. GH binds primordially to the hGH receptor and acts as a somatogen, whereas PRL and hPL bind to the prolactin receptor (PRL-R) and act as lactogens. PRL-R is a member of the cytokine receptor superfamily which presents 3 structural regions: an extracellular ligand-binding domain, a hydrophobic transmembrane domain, and an intracellular signaling domain. Multiple promoters and alternative splicing of the
PRLR gene generate several isoforms which vary exclusively in their intracellular domains and potential recruitment of signaling mediators
[90]. After ligand binding, PRL-R dimerizes which leads to the trans-phosphorylation of tyrosine residues present in Janus kinase 2 (JaK-2). This is followed by recruitment of signal transducers and transcription activators (STATs) -1, -3, or -5 which dimerize and migrate to the nucleus to enhance the expression of PRL-dependent genes
[91]. During pregnancy, the binding of hPL or PRL to the long isoform of PRL-R in pancreatic β-cells activates the JaK-2/STAT-5 pathway which results in metabolic adaptations of these cells characterized for higher transcription of GLUT-2, glucokinase, insulin, survivin, cyclin D2, and Bcl6 genes
[92]. GLUT-2 favors glucose uptake by β-cells, then glucose is phosphorylated by glucokinase and enters glycolysis/Krebs cycle/oxidative phosphorylation to increase ATP production. Higher ATP/ADP ratio blocks ATP-sensitive potassium channels, K
+ accumulation depolarizes β-cells, and voltage-gated calcium channels become activated. The resultant rise in intracellular Ca
2+ triggers insulin secretion
[93]. Additionally, transcription of survivin, cyclin D2 and Bcl6 genes increases cell mitotic divisions, avoids apoptosis, and stimulates the expansion of pancreatic islets during normal pregnancy
[94]. Beta-cell proliferation is also dependent on the downstream serotoninergic effect of both, PRL and hPL
[95]. hPL is more potent than PRL to increase insulin secretion and β-cells proliferation, whereas GH has lower potency
[96][97]. We now know that the morphologic changes in pancreatic β-cells related to pregnancy occurs largely through hPL and PRL action, but Hepatic Growth Factor (HGF), Epidermal Growth Factor (EGF), vitamin D, progestins and estrogens are also implicated
[98][99][100][101] (see
Figure 2).
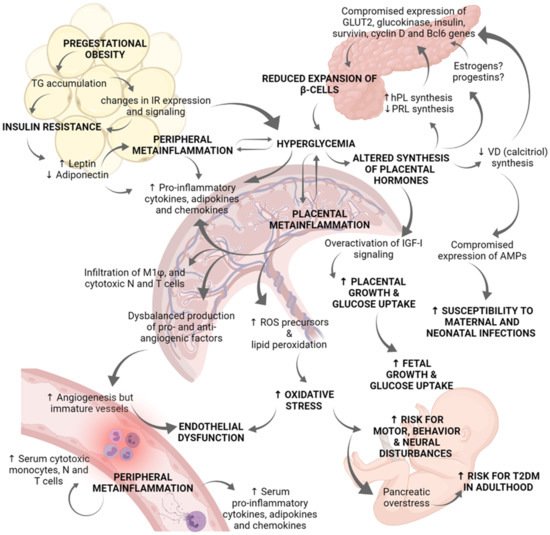
Figure 2. Role of placenta in the immunoendocrine dysregulations occurring in gestational diabetes mellitus. Hyperglycemia during pregnancy compromises the correct physiology of diverse organs, and particularly the placenta. Pregestational obesity is characterized by an excessive TG accumulation and changes in IR expression and signaling in adipose tissue, as well as in skeletal muscle cells. These changes in insulin-dependent tissues lead to insulin resistance. Then, adipose tissue secretes high levels of leptin and inhibits those of adiponectin. Leptin activation of JaK-2/STAT 3/5 pathway results in increased cytokine production contributing to peripheral metainflammation. Due to insulin resistance and altered IR signaling, women suffer chronic hyperglycemia. Clinical and biomedical studies indicate there is a positive regulatory loop between hyperglycemia and metainflammation, in which hyperglycemia induces placental and peripheral synthesis of pro-inflammatory cytokines, chemokines, and adipokines. Metainflammation also alters GLUT and IR expression which worsen hyperglycemia status. The synthesis of placental hormones is altered by hyperglycemia. Deficient synthesis of PRL and calcitriol (the active form of vitamin D) has been reported in GDM placentae, whereas hPL synthesis is increased. These placental hormonal changes, and probably estrogens and progestins, compromise pancreatic gene expression of GLUT2, glucokinase, insulin, survivin, cyclin D2, and Bcl6; all these genes are related to b-cells proliferation and survival. Because of reduced b-cells expansion, hyperglycemia worsens. Additionally, deficient synthesis of calcitriol abates placental expression of antimicrobial peptides related to innate defense, increasing mother and fetus vulnerability to infections in the perinatal period. Another endocrine dysregulation is derived from IGF-I overactivation in the placenta, which explains increased placental growth and glucose uptake. The establishment of a chronic inflammatory milieu in the placenta results in: (i) Increased production of pro-inflammatory cytokines, chemokines, and adipokines; (ii) Increased placental infiltration of M1 macrophages, cytotoxic neutrophils, and T cells; (iii) Deregulated production of pro-angiogenic and anti-angiogenic factors; and (iv) Increased lipid peroxidation and synthesis of ROS precursors. Even if angiogenesis is induced, vessels in the placenta are thickened and immature. Overall, altered vessels formation, hyperglycemia, and oxidative stress induce endothelial dysfunction. Furthermore, the serum inflammatory profile is evidenced by high levels of pro-inflammatory cytokines, chemokines, and adipokines as well as a higher presence of cytotoxic monocytes, neutrophils, and T cells. Hyperglycemia and over-activation of IGF-I signaling results in increased fetal growth which may contribute to macrosomia. Finally, hyperglycemia and oxidative stress produce pancreatic overstress in the fetus, causing an increased risk for T2DM development later in life. Experimental and clinical evidence indicates that GDM fetuses present a marked neural pro-oxidative environment which may lead to neural, motor and behavior disturbances. AMPs: antimicrobial peptides; GDM: Gestational diabetes mellitus; GLUT: Glucose transporter; hPL: human placental lactogen; IGF-I: Insulin-like Growth Factor 1; IR: Insulin receptor; N: Neutrophils; PRL: Prolactin; ROS: Reactive oxygen species; T: T lymphocytes; T2DM: type 2 Diabetes mellitus; TG: triglycerides; VD: vitamin D.
Pregnancy is considered a physiologically hyperprolactinemic state, in where PRL potentiates glucose-stimulated insulin secretion and β-cell mass. However, exacerbated hyperprolactinemia, as occurs with a prolactinoma, is related to insulin resistance
[102]. In GDM, blood levels of hPL are higher than in normal pregnancies and correlate with increased placental weight, macrosomia, hyperglycemia, insulin resistance, and altered values in an Oral Glucose Tolerance Test (OGTT)
[103][104]. In contrast, GDM mothers present similar or even lower levels of PRL than normal pregnant women
[105]. This relative contradiction seems to indicate that a delicate balance of PRL and hPL during pregnancy is needed to achieve adequate pancreatic β-cells proliferation and to avoid insulin resistance
[106]. In a diabetic mouse model, low- and high- PRL treatment induced β-cell proliferation; however, low PRL levels reduced hepatic insulin resistance whereas high PRL exacerbated it and elevated apoptosis of β-cells
[107]. A DNA sequencing study also supports the critical role of PRL-R in the control of glucose metabolism in GDM patients. In particular, two single nucleotide polymorphisms in the
PRLR gene were associated with a 2-fold risk for developing GDM
[103]. The study of physiological control of pancreatic β-mass expansion by lactotroph hormones still needs to be enlarged in the context of GDM and their co-interactions with estrogens/progestins hormones, inflammation, and obesity.
 +1 point
+1 point