Stroke is a major cause of death worldwide, leading to serious disability. Post-ischemic injury, especially in the cerebral ischemia-prone hippocampus, is a serious problem, as it contributes to vascular dementia. Many studies have shown that in the hippocampus, ischemia/reperfusion induces neuronal death through oxidative stress and neuronal zinc (Zn2+) dyshomeostasis. Glutathione (GSH) plays an important role in protecting neurons against oxidative stress as a major intracellular antioxidant. In addition, the thiol group of GSH can function as a principal Zn2+ chelator for the maintenance of Zn2+ homeostasis in neurons. These lines of evidence suggest that neuronal GSH levels could be a key factor in post-stroke neuronal survival. In neurons, excitatory amino acid carrier 1 (EAAC1) is involved in the influx of cysteine, and intracellular cysteine is the rate-limiting substrate for the synthesis of GSH. Recently, several studies have indicated that cysteine uptake through EAAC1 suppresses ischemia-induced neuronal death via the promotion of hippocampal GSH synthesis in ischemic animal models.
1. Introduction
Stroke is one of the most frequent causes of death worldwide and a major cause of serious disability. Lifestyle factors that have been shown to increase the risk of stroke include smoking; lack of physical activity; being overweight; and increased intake of salt, alcohol, and fat. These factors also impact on outcomes in patients with stroke. According to a systematic analysis from 2016, there were 80.1 million prevalent cases of stroke, 5.5 million deaths, and 116.4 million disability-adjusted life years resulting from stroke globally that year
[1]. In recent years, although the rate of deaths due to acute ischemic stroke has declined because of the introduction of new therapeutic strategies and specialized care, ischemic stroke survivors continue to suffer from disabilities
[2]. In particular, ischemia-induced damage to the hippocampus, the main brain structure related to learning and memory
[3], is recognized as a serious problem, as it leads to cognitive impairment
[4].
The pathophysiological mechanisms underlying ischemia-induced hippocampal damage have been studied intensively. Oxidative stress, a hallmark of brain ischemia, has been identified as a major factor. It results from the increased generation of reactive oxygen species (ROS) and reactive nitrogen species, which lead to apoptosis and necrosis of neurons
[5][6]. Another factor is the disruption of zinc (Zn
2+) homeostasis in the hippocampus. Zn
2+ levels are maintained at a constant level in the adult mammalian brain, including the hippocampus, in which Zn
2+ participates in normal physiological brain functions, such as learning and memory
[7]. However, ischemia/reperfusion disrupts neuronal Zn
2+ homeostasis, resulting in neuronal cell death within the hippocampus
[8]. Therefore, not only oxidative stress but also the disruption of neuronal Zn
2+ homeostasis are recognized as important therapeutic targets for the treatment of ischemia-induced hippocampal injury.
Glutathione (GSH) is the most abundant low molecular weight thiol compound within cells, and it plays a crucial role in cell defense not only against oxidative stress but also against the disruption of Zn
2+ homeostasis. In neurons, GSH levels are predominantly regulated by excitatory amino acid carrier 1 (EAAC1), which can transport cysteine, a substrate for GSH synthesis. Accumulating evidence indicates that EAAC1 gene deletion exacerbates ischemia-induced hippocampal damage via impaired Zn
2+ homeostasis as well as oxidative stress
[9], and that diurnal fluctuations in EAAC1 levels affect susceptibility to ischemia-induced neuronal cell death in the hippocampus
[10]. These findings suggest that neuronal GSH levels regulated by EAAC1 are crucial in this context.
2. Protective Roles of Glutathione (GSH) in Ischemia-Induced Hippocampal Injury
2.1. Anti-Oxidative Role of GSH in Ischemia-Induced Oxidative Stress
Cerebral ischemia leads to a marked increase in oxidative stress resulting from the generation of ROS, such as superoxide, hydrogen peroxide, and hydroxyl radicals
[5]. Oxidative stress can rapidly damage many components of the cell, including lipids, proteins, and DNA, whereby it contributes to the progression of neuronal cell death after ischemic stroke
[11][12][13]. Therefore, oxidative stress is recognized as one of the major mediators of cerebral ischemia/reperfusion-induced hippocampal injury
[6][14][15].
GSH is a major endogenous component of the cellular antioxidant defense. It is capable of scavenging various ROS directly. Research has demonstrated that the order of GSH reactivity towards the radicals is
•OH >
•OCH
3 >
•OOH >
•OOCCl
3 >
•OOCHCH
2 >
•OOCH
3, and that the rate constants range from 2.02 × 10
4 M
−1 s
−1 to the diffusion limit (7.68 × 10
9 M
−1 s
−1)
[16]. These findings indicate that GSH is an excellent free radical scavenger. On the other hand, GSH is also known to act as a substrate for a number of glutathione peroxidases to detoxify ROS
[17]. During these reactions, GSH is oxidized and converted into glutathione disulfide (GSSG), which is subsequently reduced to GSH by glutathione reductase
[18]. However, cellular GSH levels can be lowered by the export of GSSG when the production of the latter exceeds the capacity for recycling to the former, indicating that this antioxidative defense system cannot offset oxidative stress
[19][20]. Indeed, many studies have demonstrated that the brain content of endogenous GSH is depleted during ischemia/reperfusion
[21][22][23], and that treatment with GSH or glutathione monoethyl ester, a cell-permeable derivative of GSH, ameliorates ischemia-induced oxidative damage, including DNA and lipid oxidation in the hippocampus
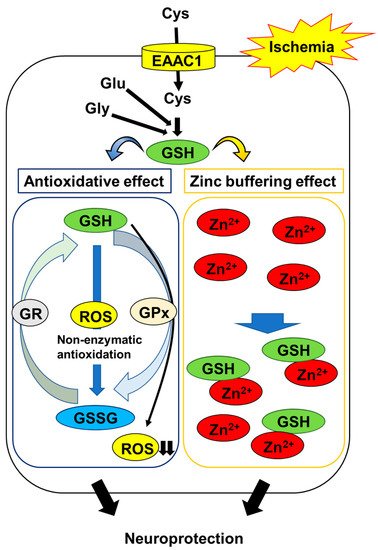
[24][25]. These observations suggest that maintaining neuronal GSH levels is an important constituent in protecting neurons against oxidative stress in the post-ischemic hippocampus (
Figure 1).
Figure 1. Mechanisms of GSH protection against hippocampal neuron damage following brain ischemia. The abbreviations are as follows: cysteine (Cys), glutamate (Glu), glycine (Gly), excitatory amino acid carrier 1 (EAAC1), glutathione (GSH), glutathione disulfide (GSSG), GSH reductase (GR), GSH peroxidase (GPx), reactive oxygen species (ROS).
Interestingly, Won et al. demonstrated that the GSH levels in CA1 pyramidal neurons were decreased during the first few hours of ischemia/reperfusion, accompanied by increased superoxide levels; conversely, the prevention of ischemia/reperfusion-induced increase in superoxide production using an inhibitor of nicotinamide adenine dinucleotide 3-phosphate oxidase, a major source of ROS, was found to suppress the decline in GSH in post-ischemic neurons
[26]. On the other hand, they also showed that mice in which neuronal GSH levels were maintained by treatment with N-acetyl cysteine (NAC), a cell-permeable precursor of GSH, exhibited a reduction in neuronal oxidative stress and neuronal cell death in the hippocampus following ischemia/reperfusion
[26]. These findings indicate that the depletion of neuronal GSH is both a result and a cause of neuronal oxidative stress following ischemia/reperfusion. In contrast, changes in the levels of GSH in post-ischemic glial cells, such as astrocytes and microglia, remain to be elucidated. Astrocytes play an important role in the synthesis of neuronal GSH by supplying precursor molecules
[27]. Therefore, it is necessary to clarify the role of glial GSH in neuronal survival in the post-ischemic hippocampus.
2.2. GSH Protects Neurons from Ischemia-Induced Disruption of Intracellular Zn2+ Homeostasis
Zn
2+ is involved in hippocampal injury following brain ischemia. In the mammalian brain, most of the Zn
2+ is bound to specialized Zn
2+ proteins—for example, metallothionein and transcription factors
[28]. A smaller part of it is concentrated in the presynaptic vesicles in a subset of glutamatergic neurons throughout the forebrain, especially in the hippocampus
[29][30][31]. Therefore, the intracellular free Zn
2+ concentration in the hippocampus is considered to be very low under normal physiological conditions
[28]. However, the stored Zn
2+, along with the protein-bound portion, is massively released during ischemia and the onset of reperfusion, subsequently accumulating within hippocampal neurons
[32][33][34][35][36][37][38]. The prevention of this process using Zn
2+ chelators, including calcium disodium ethylenediamine tetraacetate and clioquinol, has been shown to protect hippocampal neurons from ischemia-induced neurodegeneration
[8][39], suggesting that the ischemia-induced disruption of neuronal Zn
2+ homeostasis contributes to neurodegeneration in the hippocampus.
The thiol moiety of GSH has a high affinity for Zn
2+ through non-enzymatic conjugation. Treatment with GSH protects neurons against Zn
2+ accumulation-induced cell toxicity
[40]. The replenishment of GSH using NAC reduces Zn
2+ accumulation and neurodegeneration in the post-ischemic hippocampus
[41]. These findings suggest that GSH acts as an intrinsic factor that buffers intracellular Zn
2+ concentrations to prevent the ischemia-induced disruption of Zn
2+ homeostasis; in addition to its antioxidative activity, this mechanism protects neurons (
Figure 1).
In contrast, Zn
2+ binds and inhibits glutathione reductase and peroxidase, the major enzymes of the glutathione redox cycle
[42][43]. Chen and Liao demonstrated evidence indicating that Zn
2+ entry into neurons causes a decrease in intracellular GSH levels and eventually triggers neuronal cell death
[40]. GSH can suppress the ischemia-induced disruption of Zn
2+ homeostasis, but this function may be affected by the accumulation of Zn
2+ in neurons.
 +1 point
+1 point