1. Introduction
Alzheimer’s disease (AD) is a neurodegenerative disorder characterized by the progressive and inescapable cognitive deterioration, whose main neuropathological hallmarks are the formation of intracellular neurofibrillary tangles and extracellular senile plaques, producing synaptic dysfunction, and ultimately, neural death. While neurofibrillary tangles are composed of hyperphosphorylated tau protein, senile plaques are associated with neurotoxicity and are formed by aggregates of the β-amyloid peptide (Aβ), stemming from the misprocessing of the Amyloid Precursor Protein (APP)
[1][2][3]. To date, the molecular mechanisms underlying AD are not completely understood. McGeer and Eikelenboom (1994) were the first to clearly identify the inflammatory component in AD, as recently confirmed by Shippy et al. (2020)
[4][5][6]. Indeed, several authors pointed out the ability of Aβ aggregates to activate the microglial cells, thus inducing the release of inflammatory mediators such as ROS, nitric oxide, and interleukins, all responsible for neuronal death
[7][8][9][10]. Moreover, the postmortem examination of Alzheimer’s patients has confirmed the incidence of high levels of pro-inflammatory cytokines, chemokines, and inflammation mediators
[11].
Microglial cells play a central role in maintaining brain homeostasis and are involved in resolving inflammation from trauma or infectious microorganisms by means of phagocytosis and/or anti-inflammatory mediators. However, while the initial inflammatory response can be neuroprotective for the brain, the persistence of a challenging stimulation and the resultant strong microglia activation may turn the first line of defense (e.g., released cytokines) into detrimental and auto-toxic reactions that lead to synaptic dysfunction and neuronal cell death.
To date, no drugs used in anti-AD therapy seem to improve the prognosis. Indeed, the current therapeutic strategies are aimed at symptoms alleviation or the slowing down of disease progression. Hence, there is the need to introduce innovative drugs capable of interfering with the pathophysiological mechanisms underlying AD. In this scenario, growing interest has been focused on the endocannabinoid (eCB) system that is viewed as a pro-homeostatic and pleiotropic signaling system activated as an adaptive response to multiple pathological conditions
[12][13]. The eCB system consists of cannabinoid type-1 (CB
1) and type-2 (CB
2) receptors, endogenous lipid ligands such as
N-arachidonoylethanolamine (AEA) and 2-Arachidonoyglicerol (2-AG), as well as proteins and enzymes involved in their biosynthesis and inactivation. The eCB system is also considered as part of a mechanism that may operate morphological
[14], phenotypic, and functional changes of microglia and counteract the neuroinflammatory processes occurring in neurodegenerative diseases
[15][16].
Studies performed both in vitro and in vivo have highlighted CB
1-2 receptor-mediated inhibition of Aβ-induced neurotoxicity, gliosis, neuroinflammation, and memory deficits
[17][18][19]. Moreover, the pharmacological activation of CB
1 and/or CB
2 receptors improves memory and/or cognitive impairments in both AD-like transgenic and pharmacological (e.g., via intracerebral Aβ infusion) mice models
[18][20][21]. In addition, the increased expression of CB
2 levels in astrocytes- and microglia-associated neuritic plaques strongly suggest a CB
2-mediated neuroprotective effect via the stimulation of microglial proliferation and migration
[18][22].
In the past few years, several lines of evidence have suggested that eCB degradative enzymes could be a target for the development of anti-AD therapeutic options. Both the expression and activity of fatty acid amide hydrolase (FAAH), the enzyme responsible for AEA degradation, are increased in astrocytes and microglia-associated neuritic plaque from post-mortem AD patients’ brains
[17]. A reduction of AEA levels was also reported in the midfrontal and temporal cortex, where AEA levels inversely correlated with Aβ
1-42 content
[23]. Furthermore, the pharmacological modulation or genetic ablation of FAAH exerted different beneficial effects in terms of Aβ accumulation, neuroinflammation, and cognitive decline
[24][25][26]. Recently, Tanaka et al., (2019) reported that the pharmacological inhibition or ablation of FAAH in BV-2 microglia cells reduced the LPS-induced release of inflammatory mediators and pro-inflammatory cytokines, and siRNA FAAH BV-2-transfected cells led to the reduced expression of pro-inflammatory genes. These findings suggest that genetic suppression and/or pharmacological inhibition of FAAH might modulate microglial phenotypes and produce anti-inflammatory effects via different mechanisms
[27].
Taken together, these studies prompted us to investigate the effects produced by FAAH inactivation on microglial inflammatory processes. From among the different FAAH inhibitors developed so far, we selected the 3-(3-carbamoylphenyl)-phenyl N-cyclohexylcarbamate (URB597), as it is one of the best known compounds with a high affinity to FAAH. Hence, we assessed URB597 efficacy in terms of its ability to modulate morphological and functional changes in Aβ25–35-induced microglial activation in BV-2 cells. Our results show that URB597 treatment exerts a potent anti-inflammatory action by inhibiting BV-2 cell microglial polarization, by promoting cytoskeleton reorganization via cell migration and phagocytosis processes as well as by modulating the expression of pro- and anti-inflammatory markers.
2. Fatty Acid Amide Hydrolase (FAAH) Inhibition Modulates Amyloid-Beta-Induced Microglia Polarization
2.1. Aβ25–35 Induces Upregulation of the Iba1 Microglia Marker
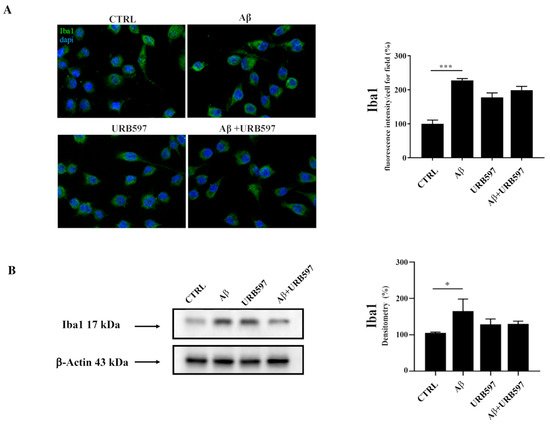
Since Iba1 is a selective marker of microglial activation following nerve injury, central nervous system ischemia, inflammatory conditions, and several other forms of brain damage, we assessed whether Aβ
25–35 could induce microglia activation by immunofluorescence and western blot analyses
[28][29]. We observed an upregulation of Iba1 in BV-2 cells treated with 30 µM of Aβ
25–35 for 24 h, thus confirming microglial activation. URB is able to decrease Aβ
25–35-induced microglia overactivation (
Figure 1).
Figure 1. (A) Immunofluorescence analysis of Iba1 in BV-2 cells treated with 30 μM Aβ25–35 for 24 h. Quantization of the intensity of the fluorescence signal was performed using the ImageJ software. The results are expressed as the mean ± SD of three independent experiments. *** p < 0.001. Bar 20 μm; (B) Determination of Iba1 protein levels by western blot. The data were normalized to the β-actin signal, reported as percentage versus control (CTRL). Data are expressed as the mean ± SEM of three independent experiments. * p < 0.05.
2.2. URB597 on BV-2 Cell Viability
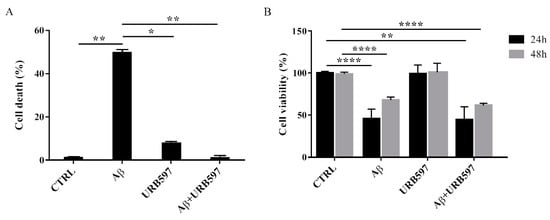
To determine the effect of Aβ25–35 in the presence or absence of URB597 on BV-2 cell viability, the Trypan blue assay and MTT analysis were performed. We selected 5 µM of URB597 concentration based on the dose-response curves on BV-2 cells (data not shown). Microglia cells were pre-treated with 5 μM URB597 and incubated with 30 μM Aβ25–35 for 24 h. Figure 2A shows a significant increase of cell death in the presence of Aβ25–35 as compared to control cells, whereas URB597 reversed Aβ25–35-induced cell death. URB597 alone had no effect.
Figure 2. Effects of Aβ25–35 in the presence or absence of URB597 on BV-2 cells. (A) Trypan blue exclusion test. Cell count was determined in BV-2 cells exposed for 24 h to 30 µM of Aβ25–35 in the presence or absence of 5 µM of URB597 and expressed as cell death (cell death/cell death + living cell). Data are reported as percentage versus CTRL. (B) Analysis of cell viability was evaluated by MTT assay. MTT reduction was analyzed in the same samples of treated cells at 24 h and 48 h. Data are expressed as percentage versus CTRL. The values are the mean ± SEM of triplicate determination from independent experiments. * p < 0.05, ** p < 0.01, **** p < 0.0001.
In addition, we performed an MTT assay using cells treated as described above. The data obtained show that Aβ25–35 induced a decrease in cell viability by around 40% at 24 h, while a slight proliferative effect of Aβ25–35 was observed at 48 h. Treatment with URB597 alone did not interfere with cell survival and did not prevent Aβ25–35 challenge at the mitochondrial level (Figure 2B).
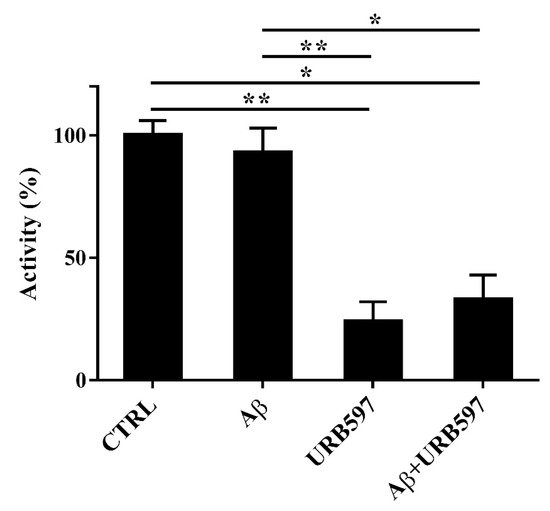
2.3. Aβ25–35 and URB597 Effects on FAAH Enzyme Activity
Since FAAH is a selective eCB-degrading enzyme, we investigated whether Aβ
25–35 and URB597 might modulate its activity. For this purpose, BV-2 cells were pre-treated with 5 μM URB597 for 4 h and incubated with 30 μM Aβ
25–35 for 24 h. Notably, Aβ
25–35 induces neurotoxicity and neuroinflammation, comparable to a full-length Aβ
1-42 peptide
[30].
Aβ25–35 did not have any significant effect on FAAH activity at 24 h (Figure 3). URB597 was also able to inhibit FAAH activity in the presence of Aβ25–35.
Figure 3. FAAH activity in BV-2 cells pre-treated with 5 μM URB597 for 4 h and incubated with 30 μM Aβ25–35 for 24 h. Data are reported as mean ± SEM of three independent experiments. * p < 0.05, ** p < 0.01.
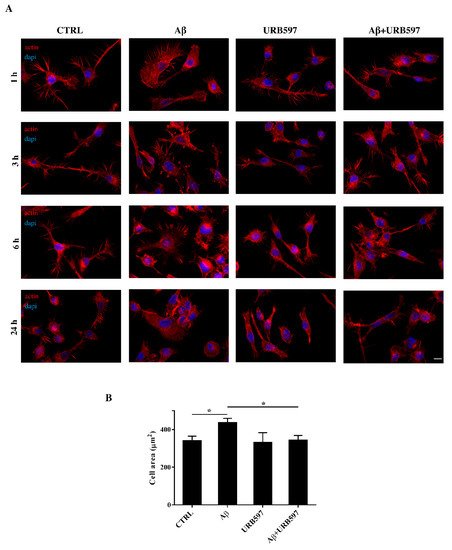
2.4. URB597 Reverts Morphological Changes Induced by Aβ25–35
Microglia rapidly responds to brain injury and disease by altering its morphology and adjusting its phenotype towards an activated state. In the activated state, microglia shifts from its resting form to an ameboid form (M1/M2)
[31]. Cells increase their surface area and acquire a flat morphology. To evaluate the effects of FAAH inhibition on cell morphology, we performed an immunofluorescence analysis through the phalloidin staining of F-actin on BV-2 cells pre-treated with 5 μM of URB597 for 4 h and incubated with 30 μM of Aβ
25–35 (
Figure 4). Cell morphology was analyzed at different time points (1 h, 3 h, 6 h, 24 h).
Figure 4. (A) Analysis of BV-2 cell morphology by rhodamine-conjugated phalloidin (TRITC-phalloidin) staining to highlight actin and 4′, 6-diamidino-2-phenylindole (DAPI) to detect the nucleus, after pre-treatment with 5 μM URB597 for 4 h and incubated with 30 μM Aβ25–35 for 1 h, 3 h, 6 h, and 24 h. (B) Cell areas were quantified at 24 h using Image J software. Data were reported as mean ± SD of at least three independent experiments. * p < 0.05. Bar: 20 µm.
The results show that control cells exhibited a very small cell body. They had long cell ramifications and actin was organized predominantly in the filopodia, used by the cells to explore the surrounding environment. At all time-points analyzed, cells treated with Aβ25–35 increased their surface area, acquiring a flat and polygonal morphology. Furthermore, cells retracted the branched processes that is typical of microglia in its resting form. On the other hand, cells treated with URB597 had a more rounded morphology, characterized by the shrinkage of the cellular body and the presence of several cellular processes. BV-2 cells co-treated with URB597 and Aβ25–35, showed a phenotype similar to that obtained with only URB597 (Figure 4A). These results were also confirmed by the quantitative analysis carried out by measuring the cellular area expressed in μm2 at 24 h. The quantitative analysis confirmed that the treatment with URB597 alone produced surface areas similar to those of control cells, while after stimulation with Aβ25–35 the cells underwent an enlargement of their soma. The cell had an area of 440 μm2 while the control sample was at 340 μm2. In the combined treatment, URB597 reduced the area to values comparable to those of controls, indicating the capacity of URB597 to restore the amoeboid phenotype observed in the presence of Aβ25–35 (Figure 4B).
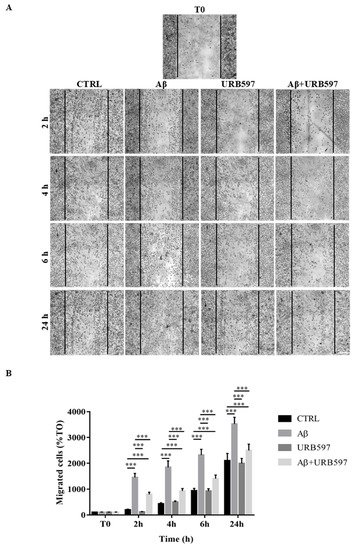
2.5. Effect of URB597 on Cellular Migration
One of the aspects of microglia activation is the acquisition of a migratory phenotype, which is essential for the cells to move towards the insult site. In this study, we performed a scratch assay in order to evaluate URB597-induced migratory capacity. The cells were pre-treated with 5 µM URB597 for 4 h in the presence or absence of 30 µM Aβ25–35 for 2 h, 4 h, 6 h, and 24 h. Migration was quantified by counting the cells that migrated from the border of the scratch to the uncovered areas, and the values obtained were reported on the graph (Figure 5B).
Figure 5. (A) Analysis of cell migration determined by scratch assay. BV-2 were pre-treated with 5 µM URB597 for 4 h in the presence or absence of 30 µM Aβ25–35 for 2 h, 4 h, 6 h, and 24 h. T0 represents the control at time 0, and CTRL the control for each time point. (B) The results were normalized vs. T0 sample and re-ported as percentage. They represent the mean ± SD of three independent experiments. *** p < 0.001. Bar: 200 μm.
As shown in Figure 5, treatment with Aβ25–35 induced migration just 2 h after stimulation, as compared to the control cells. We demonstrated that URB597 alone did not affect cell migration, and that URB597 pre-treatment reduced BV-2 migration induced by Aβ25–35. The migratory effect was directly proportional to the number of incubation hours.
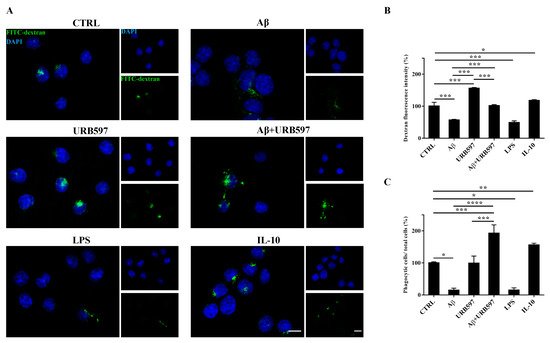
2.6. URB597 Increases Microglia Phagocytic Capacity
To determine whether URB597 has a role in phagocytosis, we assessed FITC-dextran microglial uptake. BV-2 cells pre-treated with 5 µM URB597 for 4 h and incubated with 30 µM Aβ
25–35 for 24 h were incubated for 1 h with FITC-dextran and analyzed by a microscope; the fluorescence images are shown in
Figure 6A. As shown in
Figure 6B, we evaluated the intensity of the fluorescent dextran in the different treatments. BV-2 cells treated with Aβ
25–35 showed a reduced ability to absorb dextran compared to untreated cells. The sample treated only with URB597 showed an intensity value comparable to that obtained by IL-10 stimulation. In the combined treatment with Aβ
25–35 and URB597, the inhibitor turned out to be capable of increasing the intensity value as compared to the Aβ
25–35 sample. LPS and IL-10 were used as reference compounds that could reduce
[32] or increase phagocytosis
[33][34]. Moreover, the number of dextran-positive cells was quantified on the total cells and the results were reported as percentage (
Figure 6C). After stimulation with Aβ
25–35 as well as with LPS, the number of dextran-positive cells was reduced by approximately 80% relative to the control cells. On the other hand, treatment with URB597 alone showed values similar as the control but lower if compared to the IL-10 treated sample. In the Aβ
25–35 and URB597 combined treatment, the inhibitor improved the number of dextran-positive cells, with higher values of phagocytic cells as compared with the Aβ
25–35 treated sample.
Figure 6. Analysis of the phagocytosis process using FITC-dextran on BV-2 cells pre-treated with 5 μM URB597 and incubated with 30 μM Aβ25–35 for 24 h. (A) Representative immunofluorescence images of FITC-dextran uptake. (B) Quantization of the intensity of FITC-dextran. (C) Quantization of the number of dextran-positive cells on the total cells. The data were normalized to CTRL and reported as a percentage. The results were expressed as the mean ± SD of three independent experiments. Statistical significance was determined using ANOVA analysis by Tukey’s test. * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001. Bar: 20 µm.
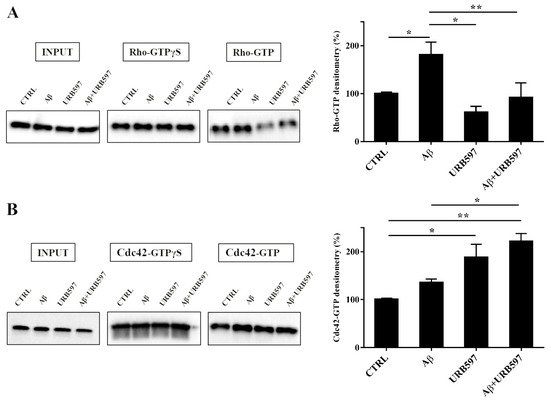
2.7. URB597 Affects Rho GTPases Activity
Cytoskeleton plasticity and the formation of actin-rich structures, with consequent morphological modifications, are regulated by the Rho GTPase protein family. Since the Rho GTPase family is a master regulator of cytoskeletal reorganization and plays an important role in membrane trafficking
[35][36], we investigated whether the Rho and Cdc42, components of the Rho GTPase family, were able to regulate the different activated states of microglia cells. To evaluate the influence of the FAAH inhibitor on this class of proteins, we performed pull-down analyses on the total protein extraction from BV-2 cells pre-treated with 5 μM URB597 for 4 h and incubated with 30 μM Aβ
25–35 for 24 h. We examined RhoA, which is involved in the assembly of contractile actin, and Cdc42 proteins that is involved in filopodia formation.
This analysis allows for the precipitation of active GTPases proteins through a specific binding protein. Our results show a marked increase of RhoA activity in the samples treated with Aβ25–35 as compared to control cells, whereas a decrease was observed in the URB597-treated samples with respect to Aβ25–35 samples (Figure 7A). As shown in Figure 7B, Cdc42 activity significantly increased in samples treated with URB597, considering both against Aβ25–35 and control cells samples.
Figure 7. Pull-down assay of Rho and Cdc42 proteins (A,B). BV-2 cells were pre-treated with 5 μM URB597 for 4 h and incubated with 30 μM Aβ25–35 for 24 h. The pull-down assay with GTPγS was used as positive control. The results are reported as a percentage versus control (CTRL). Densitometric analyses were performed with ImageLab software (Biorad) and normalized to INPUT. * p < 0.05, ** p < 0.01.
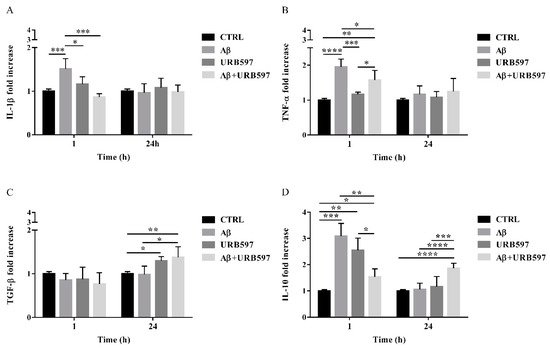
2.8. URB597 Reduces mRNA IL-1β and TNF-α Expression, Increases TGF-β and IL-10 Expression
The activation of Rho GTPases supports the hypothesis of Aβ-driven cytoskeleton rearrangement, ultimately leading to an increase of migration and a reduction of phagocytosis. Since these effects were counteracted by URB597, we aimed at determining the level of inflammation in our BV-2 cell model.
The expression of pro-inflammatory cytokines such as IL-1β and TNF-α as well as anti-inflammatory cytokines such as TGF-β and IL-10, were analyzed by qPCR. BV-2 cells were pre-treated with 5 µM URB597 for 4 h and incubated with 30 µM Aβ25–35 for 1 h and 24 h. The results presented in Figure 8A,B demonstrate that IL-1βand TNF-α expressions increased within 1 h in BV-2 cells treated with Aβ25–35 as compared to the control. On the contrary, a reduction of cytokines was observed in the sample with the combined treatment as compared to Aβ25–35 alone. No changes were detected for IL-1β and TNF-α levels in BV-2 cells incubated with 30 µM Aβ25–35 for 24 h. We also observed that treatment with Aβ25–35 did not modify TGF-β expression, while URB597 alone or in combination with Aβ25–35 upregulated TGF-β at 24 h, as shown in Figure 8C.
Figure 8. The mRNA expression of different inflammatory cytokines such as IL-1 β (A), TNF-α (B), TGF-β (C), and IL-10 (D), monitored by qPCR and normalized to 18S ribosome subunit. Data are shown as mean ± SD from three independent experiments performed in triplicate. Expression profiles were determined using the 2−ΔΔCT method. * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001.
Moreover, both Aβ25–35 and URB597 administration, alone or in combination, induced an increase of IL-10 expression within 1 h, while after 24 h of stimulation the effect was persistent only in the Aβ25–35 and URB597 combined treatment (Figure 8D).
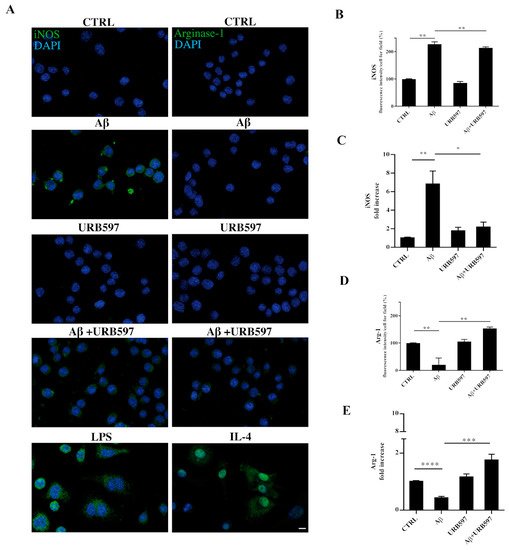
2.9. URB597 Modulates Both iNOS and Arg-1 Expression
The activation of microglia is a polarized process that can lead either to the potentially neurotoxic M1-activated phenotype or to the neuroprotective M2-activated phenotype
[37]. To evaluate the M1 or M2 states in the treated cells, we performed both immunofluorescence and RT-qPCR analyses and assessed the level of nitric oxide synthase (iNOS) and Arginase-1 (Arg-1), which are markers of the M1 and M2 microglia phenotypes, respectively (
Figure 9). BV-2 cells were pre-treated with 5 µM URB597 for 4 h and incubated with 30 µM Aβ
25–35 for 24 h. LPS and IL-4 were considered as positive controls for iNOS and Arg-1 markers, respectively. As shown in
Figure 9A–D, the stimulation of BV-2 cells with Aβ
25–35 induced a significant enhancement of iNOS, whereas a reduction of Arg-1 was observed in comparison to control. The combination of URB597 and Aβ
25–35 induced a decrease in iNOS expression and an increase in Arg-1 with respect to Aβ
25–35. URB597 alone did not affect the expression of both markers.
Figure 9. BV-2 cells were pre-treated with URB597 5μM for 4 h and incubated with Aβ25–35 30 μM for 24 h. LPS and IL-4 were used as positive controls for iNOS and Arg-1 markers, respectively. Representative immunofluorescence images of iNOS and Arg-1 (A). Quantitative immunofluorescence analysis of iNOS (B) and Arg-1 (D). Quantification of fluorescence signal intensity was analyzed using ImageJ software. The results are expressed as the mean ± SD of three independent experiments. ** p < 0.01. Bar 20 μm. iNOS (C) and Arg-1 (E) mRNA expressions were evaluated by qRT-PCR at 24 h. Data are shown as mean ± SD from three independent experiments performed in triplicate. Expression profiles were determined using the 2−ΔΔCT method. * p < 0.05, ** p < 0.01. *** p < 0.001, **** p < 0.0001.
 +1 point
+1 point