1000/1000
Hot
Most Recent
 +1 point
+1 point
Prion is an atypical etiological agent composed solely of a misfolded protein—(proteinaceous infectious particle), which affects mammals causing a group of slow, progressive, neurodegenerative, lethal, untreatable disorders known as transmissible spongiform encephalopathies (TSEs).
Prion (pronounced pree-on) is an atypical etiological agent composed solely of a misfolded protein — (proteinaceous infectious particle [1]), which affects mammals causing a group of slow, progressive, neurodegenerative, lethal, untreatable disorders known as transmissible spongiform encephalopathies (TSEs). Historical documentation of prion diseases dates back nearly three hundred years when a disorder referred to asscrapiewas reported in sheep [2] and later in goats. Other TSEs include bovine spongiform encephalopathy (BSE, also known as mad cow disease) in cattle; chronic wasting disease (CWD) in deer, elk, and moose; exotic ungulate encephalopathy (EUE) in nyala, oryx, and greater kudu; and related encephalopathies in camel, mink, and cat. In humans, prionic disorders include Creutzfeldt–Jakob disease (CJD), Gerstmann–Sträussler–Scheinker syndrome (GSS), fatal familial insomnia (FFI) and kuru (reviewed in [2][3]).
It is perplexing to observe that, opposed to other conventional pathogens (viroids, viruses, bacteria, fungi, and parasites) which are extrinsic to the host, the misfolded prion protein has an endogenous origin. The natively folded version of the prion protein, named cellular prion (PrPC), is a cell-surface glycoprotein encoded by the endogenous mammalian gene Prnp, which is present in all vertebrates [4] and highly expressed in the brain, and at lower levels in most other tissues. Its precise organismal/cellular function is not clear, although many roles have been proposed, including stem cell renewal [5], memory formation, and myelin homeostasis [6]. and extracellular aggregates, which are possible causes of neuronal death.
It is suggested that the physical interaction between PrPC and PrPSc (as a monomer or a fibril) suffices for the conversion of the first into the second [7]. Thus, it is interesting to highlight that pathogenic prion (PrPSc) amplification is not directly dependent on nucleic acids as all other known classes of pathogens. Therefore, pathogenic prion multiplication is an event of structural information amplification, rather than DNA or RNA replication. Nevertheless, since PrPC synthesis is dependent on translation, transcription, and ultimately on a DNA sequence, PrPSc replication is indirectly dependent on DNA.
Notoriously, in the last decade, a series of discoveries have evidenced that the ‘prion principle’ may not be limited to the prion protein itself, but rather to many other mammalian (for example, a-synuclein and ataxin) and fungal (Sup35, Ure2; reviewed in [8]) polypeptides which are able to have a prion-like behavior (“prionoids”; [6]). These proteins present two structural conformations, the first acting as a template to the second, which is aggregation-prone, pathogenic and able to amplify its structural conformation. Many of these mammalian proteins are associated with neurodegenerative disorders, such as amyloid precursor protein in Alzheimer’s disease (AD), ataxin in amyotrophic lateral sclerosis (ALS), huntingtin in Huntington’s disease (HD) and a-synuclein in Parkinson’s disease (PD) (reviewed in [9]).
MicroRNAs (miRNAs) are small (18–25 nucleotides) non-coding transcripts responsible for modulating the expression of other genes post-transcriptionally, via interaction with partially complementary sites at corresponding target RNAs. Such hybridization is mediated by a special ribonucleoprotein complex (miRISC), leading to targeted gene downregulation via RNA cleavage, deadenylation or translational inhibition (Figure 1) (reviewed in [10]). MiRNAs may also act through other mechanisms, such as transcriptional control (reviewed in [11]).
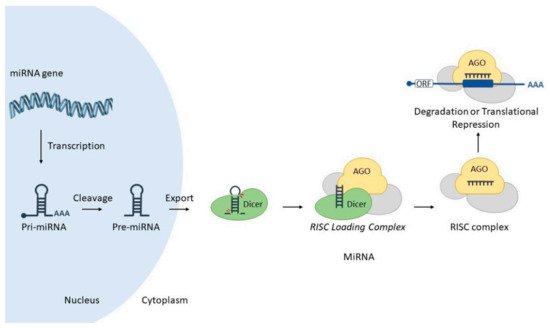
In general, miRs are intrinsically pleiotropic since one miRNA may interact with hundreds of different target RNAs, while one target RNA may also be regulated by different miRNAs. Virtually all cellular processes have been shown to be modulated by miRNAs, and since their discovery, they have been acknowledged as a new level of gene regulation, allowing the fine-tuning of genetic expression.
Since both prion protein and microRNAs are encoded by the mammalian genome, both of them are subjected to many levels of expression control, as well as inter-regulation. Here we will briefly present some aspects of such molecular interactions between both actors.
The identification of endogenous miRNAs able to downregulate prion protein has important implications for potential therapies, since decreasing PrPC hinders PrPSc amplification. Accordingly, Pease and colleagues (2019) [13] decided to investigate, in vitro, which endogenous miRNAs are able to modulate prion protein. They constructed a sophisticated high-throughput arrayed screen, using a genome-wide miRNA library (a total of 2019 miRNA mimics), reverse transfection, and time-resolved fluorescence resonance energy transfer. The other four miRNAs (miR-124-3p, miR-192-3p, miR-299-5p, and miR-376b-3p) promoted changes in PRNP protein, however, probably by interacting with indirect regulators of PrPC.
For example, in order to understand the involvement of PrPC accumulation in diabetic dementia-like pathology, Kalani et al. [14] assessed miRNAs expression via RT-qPCR array in brain cells of diabetes mellitus In addition, sequence analysis showed that miR-146a is able to bind to a conserved domain in the murinePrnpgene. Taken together, their data suggested that miR-146a modulates PRNP, and its downregulation in db/db diabetic mice explains PrPCaccumulation, previously known to be associated with the dementia-like phenotype.
In this context of miRNA-mediated post-transcriptional regulation, the identification of nucleotide variations within target sequences can provide indirect explanations for specific phenotypes. Given the strict requirement of high complementarity between a miRNA seed sequence and the 3′UTR portion of a target mRNA, nucleotide mutations within the seed sequence are typically detrimental to miRNA-mRNA interaction, hence directly affecting post-transcriptional gene regulation mechanisms. To investigate whether this inconsistency was due to post-transcriptional regulation, Zhao and colleagues (2017) [15] decided to annotate the 3′UTR region of buffalo Prnp gene by 3′ rapid-amplification of cDNA ends, and sequencing. Additionally, the authors also performed luciferase reporter assays showing the direct interactions of five miRNAs (miR-125b-5p, miR-132-3p, miR-145-5p, miR-331-3p, and miR-338-3p) in the fixed differences sites in buffalo 3′UTR PRNP, and validated such downregulations by RT-qPCR.
Some studies have revealed that a fraction of prion proteins remain in the cytoplasm, but their function in this cellular region was not clear. In order to shed light on this issue, Beaudoin and colleagues (2009) [16] created truncated versions of PrP, which tended to localize in the cytoplasm, forming a perinuclear ribonucleoprotein complex (RNP). This structure resembled the ‘chromatoid body’, an RNA granule possibly involved in post-transcriptional gene regulation, typically found in germ cells and planarian stem cells and neurons. Their observations shed light on a possible role of PRNP in the cytoplasm, as well as suggested that potential interactions between prion protein and miRNA machinery in this perinuclear RNP may impact miRNA biogenesis.
Different studies have proposed diverse roles for prion protein, such as cell signaling, adhesion, and differentiation [17]. [18] decided to investigate whether prion protein could act as a modulator of this process in adipose-derived stem cells (ADSCs), an interesting source of stem cells. They submitted ADSCs to neuronal induction with IBMX (a competitive non-selective phosphodiesterase inhibitor), and through a series of experiments, the authors observed that PRNP protein contributes to the upregulation of miRNA-124 during this differentiation process in mouse ADSCs. small C-terminal domain phosphatase 1 (SCP1) mRNA, an important anti-neural factor, thus prompting neuronal differentiation.
Interestingly, previous studies have evidenced putative links between prion protein and miRNA pathways. For example, via a protein-protein interaction screen, Satoh and colleagues (2009) [19] showed that recombinant PrPC binds AGO1 (a key protein in the RNA interference and miRNA machineries). Interestingly, although most prion protein is exposed to the extracellular milieu, one endogenous transmembrane PrPC variant presents its N-terminal ‘octarepeat’ domain in the cytoplasm. Based on this fact and other evidence, Gibbings and colleagues (2012) [20] set out to interrogate this cellular process.
MiRNAs are crucial musicians in the molecular orchestra of the cell, and their expressions are dysregulated under stress conditions, as well as in several diseases [21]. Thus, many research groups have investigated miRNA expression profiles in patients diagnosed with prion diseases [22][23][24][25] as well as in animal models, such as (i) scrapie-infected murine models [26][27][28][29][30][31], (ii) SMB-S15 cells [29], (iii) scrapie-infected murine neuroblastoma cells [32], Moreover, temporal miRNA expression studies have highlighted the phase-specificity of miRNAs and their changing patterns throughout the progression of the disease [24][27][28][33].
[26] presented, for the first time, the global expression analysis of mature miRNAs in murine brains during prion disease. In their study, mice were intracerebrally inoculated with scrapie strain 22A, and miRNA expression was determined via microarray and RT-qPCR procedures. Interestingly, the authors found several potential targets for these miRNAs through computational analyses, 119 of which had already shown to be dysregulated in murine prion disease models. They also conducted a computational-biochemical (in silicotarget prediction/dual-luciferase reporter assay) integrative approach to validate miRNA targets and found potential targets involved in signaling pathways related to cell death, synapse function, and neurogenesis [26].
The heterogeneous composition of the brain tissue, which includes different neuronal and supporting cell types (e.g., astrocytes, oligodendrocytes, and microglia), may mask temporal gene expression changes associated with prion-affected neurons. [27] used laser capture microdissection technology to isolate neurons from the CA1 hippocampus region of scrapie-infected and control mice. Using microarray analysis, they were able to unveil a well-defined cluster of differentially expressed genes during the preclinical phase, including deregulated miRNAs miR-132-3p, miR-124a-3p, miR-16-5p, miR-26a-5p, miR-29a-3p, and miR-140-5p, which were validated by RT-qPCR. (miR-132-3p, miR-124a-3p, miR-29a-3p) might help neurons evoke a pro-survival response and stimulate dendrite remodeling mechanisms at the preclinical stage of prion disease [27].
Importantly, these miRNA alterations are shown to be dynamically related to the stage of prion disease [27]. In this regard, Toivonen and collaborators (2020) [31] decided to usePrnp-transgenic Tg501 mice to explore these temporal miRNA fluctuations in preclinical and clinical stages of mouse-adapted goat scrapie infection, comparing with age-matched, mock-inoculated controls. By carrying out small RNA sequencing of the cervical spinal cord, cerebellum, and plasma, they were able to identify significantly stage-specific deregulated miRNAs in preclinical- and clinical-stage animals (three and 23 miRNAs, respectively), which were predicted to be involved within known biological pathways, such as prion disease, extracellular matrix interactions, glutaminergic synapse, axon guidance, and transforming growth factor-beta signaling [31]. However, since the preclinical stage showed minimal changes, the authors suggested that most miRNA alterations are triggered by advanced prion-associated pathology, making it challenging to use them for clinical purposes, such as diagnostic biomarkers.
[29] carried out small RNA deep sequencing to unravel miRNA expression profiles in the brains of terminal-stage mice infected with scrapie agents 139A, ME7, and S15. As a result, a total of 57, 94, and 135 differentially expressed miRNAs were identified in 139A-, ME7- and S15-infected mice, respectively, whereas 36 miRNA alterations (up- and downregulations) were common to all infected groups—including two novel miRNAs, novel-miR-2 and novel-miR-20. Additionally, the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis showed a total of 12 biological pathways shared among those three groups, including (i) olfactory transduction, (ii) metabolic pathways, and (iii) bacterial invasion of epithelial cells, which indicates a great similarity and coincidence of the potentially affected pathways in the terminal-stage brains infected with three different scrapie agents [29].
Whilst gene expression profiles were often explored in transmissible or genetically forms of prion pathologies, miRNA dysregulations associated with sporadic CJD (sCJD) remained poorly elucidated. In this regard, Llorens, Thüne and collaborators (2018) [24] carried out small RNA-Seq to investigate whether miRNA accumulation and its machinery (proteins Dicer, Drosha, microprocessor complex DGCR8, and Exportin-5) displayed differences in the two most affected brain regions (frontal cortex and cerebellum) within the two most prevalent sCJD subtypes (known as MM1 e VV2). Curiously, only 31% and 10% of the deregulated miRNAs were common to both subtypes in the frontal cortex and cerebellum, respectively, evidencing a considerable degree of divergence within sCJD. Finally, they revealed that human brain and cerebrospinal fluid (CSF) miRNA profiles did not correlate, possibly due to the difference in the period of analyses—time of diagnosis for CSF versus post-mortem for brain tissue.
Interestingly, it was observed that miRNA-146a is upregulated in human brain cells infected with at least five distinct viral species, in diverse stress-induced human neuronal-glial primary cell co-cultures [34][35], as well as in a mouse model for scrapie [26] and in AD brain [36][37]. In order to investigate the profile of miRNA-146a in sCJD Thus, the role of this miRNA, which is abundantly expressed in murine and human brains, was expanded to other prion diseases, suggesting it is an integral part of the innate immune or inflammatory brain cell responses, with wide action. Moreover, this finding, along with previous studies, evidenced a shared underlying inflammatory response to neurological insults promoted by single-/double-stranded RNA/DNA viruses, AD, and prion diseases, when compared to healthy aged human controls.
Thus, Burak and colleagues (2018) [33] decided to conduct an experimental approach in which they overexpressed this miRNA using a lentivirus in mature hippocampal neurons of mice. Interestingly, they observed that neurons with higher levels of miR-16 displayed decreased neurite length and branching [33]. Moreover, via immunoprecipitation of Ago2-containing RISC complexes followed by microarray analysis, they identified some miRNA-16 targets including TrkB (NTRK2), MEK1 (MAP2K1), and c-Raf (RAF)—members of neurotrophin receptor-mediated MAPK/ERK pathway. Taken together, induction of miR-16 and the consequent downregulation of its targets might explain reduced neurite branching and length during the presymptomatic phase of prion disease.
As previously shown, miRNA-146a was reported as an inflammation mediator molecule in neurodegenerative diseases, including scrapie. Likewise, Saba and collaborators (2012) [38] evidenced through a series of different experimental approaches (microarray, RT-qPCR, gene overexpression/knock-down) that miR-146a is upregulated in the brain tissues of a murine model of prion disease, concomitantly with the onset of PrPScdeposits and activation of microglia. Authors suggested an additional function for miR-146a as an important regulator of microglial function by modulating the activation state during prion-induced neurodegeneration [38].
For example, in prion disease, decreases in synapses and densities of the dendritic spine are observed in neurons of the cortex and hippocampus through the preclinical stage. Based on these key aspects, Boese and colleagues (2016) [28] hypothesized that miRNAs regulate synaptic protein synthesis in response to prion replication. They probed miRNA expression in synaptoneurosomes prepared from murine forebrain and hippocampus at two stages of prion disease. Authors noted that several of those miRNAs target proteins associated with synaptic function and structural morphology, findings which may help understand prion pathology.
Interestingly, Jozef Nahalka (2019) [39] investigated an innovative, yet controversial concept of “protein-RNA recognition code” in the context of several neurodegenerative diseases (AD, HD, PD, ALS) and prion disorder. H and SFPQ proteins, as well as between (CTG)n repeats in RNA, MNBL proteins, and RNA foci in myotonic dystrophy. More importantly, the code predicted that miR361 interacts with the spacer of the second and third α-helices of the prion protein, which seems to be important for prion conformational change, as well as with the ‘octapeptide repeat’ region. Accordingly, one study [28] in an animal model of prion disease reported deregulation of this microRNA, potentially evidencing a link between these two actors.
According to Montag and collaborators (2012) [32], disbalance in cholesterol homeostasis is related to the progression of neurodegenerative disorders, like prion diseases. Thus, in order to detect miRNAs involved in this process, they performed an ultra-deep RNA sequencing in scrapie-infected neuroblastoma N2a cells, including differential expression analysis against N2a-mock cells and subsequent validation by RT-qPCR. Interestingly, they found two downregulated miRNAs localized in a 5 Kb genomic cluster on the mouse X-chromosome— , that putatively bind to the 3′UTR of the cholesterogenic mouse genes involved in prion-induced dysregulation of cholesterol homeostasis Hgmcs11, Hgmcr, ldi1, CYP51, Ldlr, and Srebf2.
Intriguingly, protein misfolding processes—exemplified by the prion protein—are also manifested in highly prevalent neurodegenerative diseases such as Alzheimer’s (AD) and Parkinson’s diseases (PD) In general, the misfolding and aggregation processes in PrPC-to-PrPSc conversion are shared in PMDs [40]—β sheet-rich oligomers forming amyloid-like aggregates by a seeding-nucleation mechanism [40][41][42]. Moreover, the pathological mechanisms of prion disease transmission are also likely to be mirrored in AD, since experiments with transgenic mice expressing human amyloid protein have shown to develop the specific abnormalities of an AD profile—accelerated Aβ-deposition in brain and proteolysis-resistant seeding-competent The transmission of protein misfolding is also observed in Tau proteins (AD) [43], Lewy bodies (α-synuclein aggregates) in PD [44][45][46], which suggests the prion-like transmission behavior in PMDs (reviewed in [9]).
On the other hand, under pathological conditions these inflammatory processes become dysregulated—uncontrolled neuroinflammation—and the CNS undergoes elevated glial cell activation, BBB permeability, and peripheral immune cell infiltration. [47] depict how the aforementioned miRNAs play similar overlapping roles within multiple pathologies. For instance, miR-155 acts as a pro-inflammatory factor in a similar way in MS, AD, and PD [48][49][50][51][52][53][54][55][56]; likewise, miR-146a operates as an anti-inflammatory agent in MS, AD and conventional prion diseases [38][48][54][56][57]. Additionally, Slota and Booth (2019) also highlighted how the discovery of disease-specific miRNA signatures in circulating fluids (e.g., serum, blood, and plasma) might paves a way for the use as non-invasive biomarkers to circumvent the complicated diagnosis of symptom-overlapping neurological disorders (reviewed in [47]).
Multiple neurodegenerative diseases, such as AD, ALS, HD, PD, and conventional prion diseases share pathophysiological processes that enable us to pinpoint common molecular mechanisms across them, thereby favoring the development of novel therapeutic approaches. In the search of miRNA dysregulation patterns across neurodegenerative diseases, Juźwik and colleagues (2019) [58] decided to perform a non-biased systematic review on this topic (miRNA and neurodegenerative diseases). A total of 2318 significantly dysregulated miRNAs (validated by qPCR only) were retrieved from each of the 641 accepted manuscripts, but only seven individual miRNAs (downregulated—miR-9-5p, miR-21-5-p, miR-124-3p, miR-132-3p; upregulated—miR-146a-5p, miR-155-5p, miR-223-3p) and 1 miRNA family (miR-29 family) were considered as prevalent across neurodegenerative diseases [58]. Likewise, NF-κB signaling, BBB maintenance, neurogenesis, autophagy homeostasis, and microglial activation pathways involve the functioning of multiple miRNAs.
Finally, Brennan and others (2019) [59] performed a wide literature search on PUBMED and Google Scholar platforms in order to identify human body fluid miRNA biomarkers for AD, PD, MS, and familial and sporadic forms of ALS reported in articles published from 2011 to 2018. The result was a ‘knowledge base’ encompassing 72 different studies and 347 unique miRNAs considered statistically significant. Interestingly, the authors noted that the prion disease pathway is one of the most constantly pictured pathways in their study, with high statistical significance [59]. Finally, the authors highlight that fifty-seven percent of all miRNAs reported in other articles as dysregulated in prion disease were also identified in their human body fluid-focused survey, for example, hsa-miR-146a, miR-26a, hsa-let-7i, hsa-miR-424, and hsa-miR-128 [59].





