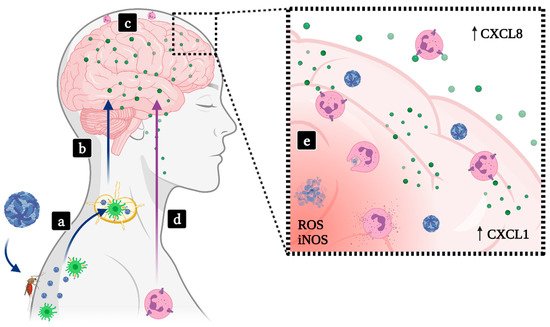
2.1. Hemorrhagic Fevers
Viral hemorrhagic fevers (VHF) are a group of illnesses that affect multiple organ systems and are characterized by high fever, increased vascular permeability, decreased plasma volume, coagulation abnormalities, shock and hemorrhage
[9]. Four different RNA viral families are known to cause VHF including
Arenaviridae,
Bunyaviridae,
Filoviridae and
Flaviviridae. Different viruses with different invading strategies and disease mechanisms can lead to VHF. Despite those differences, the severity of these individual diseases roughly correlates with the onset of vascular pathologies
[10].
YFV and DENV are two flaviviruses known to cause VHF. Both viruses are mainly transmitted by
Aedes aegypti mosquitoes and can be maintained in a sylvatic, enzootic cycle
[11]. The mosquito saliva contains molecules known to modulate host inflammation, increasing cell susceptibility to the viruses. It is suggested that mosquito bite enhancement of virus infection is not related to interferon response suppression. Instead, host increased susceptibility is closely related to neutrophil-dependent inflammation after a bite
[12][13]. Following a bite, mast cell degranulation markedly increases the expression of IL-1β, IL-6 and neutrophil-attracting chemokines such as CXCL1, CXCL2, CXCL3 and CXCL5. Neutrophils are recruited to the site and express high levels of IL-1β, which orchestrates the local inflammatory environment by leading to the recruitment of myeloid cells, a primary target cell for flaviviruses
[13] (
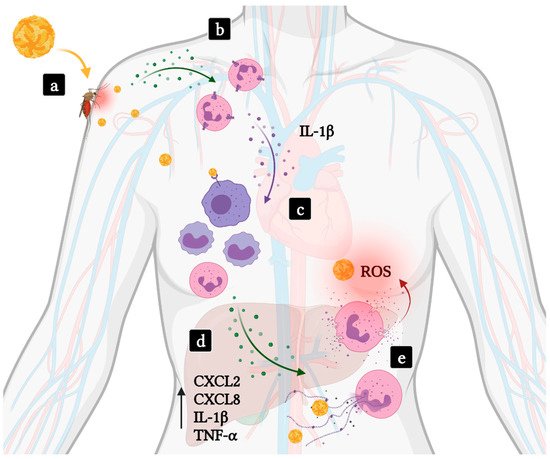
Figure 1).
Figure 1. Involvement of neutrophils in hemorrhagic fever and shock induced by flavivirus infections. (a) Viruses such as DENV or YFV (yellow viral particles) are transmitted to human hosts through an infected mosquito bite. (b) Local inflammation results in the recruitment of neutrophils to the infected site. (c) Activated neutrophils amplify inflammation secreting IL-1β. Subsequent activation and recruitment of mononuclear phagocytic leukocytes, target cells in flavivirus infections, promotes viral replication and dissemination of infection. (d) Severe disease takes place as high levels of proinflammatory cytokines are produced, including those participating in neutrophil recruitment and activation, and result in systemic inflammation and multiorgan failure. (e) Levels of ROS and MPO-DNA complexes correlate with disease severity. Green dots—Neutrophil chemoattractants; Purple dots—IL-1β.
A common underlying mechanism in VHF is a massive production of proinflammatory cytokines. High levels of cytokines such as CXCL2, CXCL8, IL-6, TNF-α are usually found in the blood of severe yellow fever and dengue fever patients
[14][15]. The role of neutrophils in severe YFV infection is unclear. However, similar to what is observed in severe DENV infections, levels of CCL2, IL-1β, IL-1Ra and TNF-α are increased
[16]. Recent evidence supports the involvement of neutrophils in the pathogenesis of severe dengue fever
[17][18]. Given the similarities between the inflammatory pathways triggered in severe cases of Dengue and Yellow fever, neutrophils may also play an important role in YFV pathogenesis. CXCL2, CXCL8, IL-1β and TNF-α participate either in neutrophil recruitment or activation, and more studies are needed to better characterize the relationship between neutrophils and disease severity.
By comparing the transcriptional signatures of patients with early, severe and mild dengue symptoms, Hoang et al. showed that a major difference exists in the abundance of neutrophil-associated transcripts in humans
[19]. Another study has shown an increase in the levels of elastase activity and other NETs components in Dengue hemorrhagic fever patients (DHF) when compared to patients presenting with mild disease, suggesting an association between neutrophil activation with disease severity. DENV infection leads to the production of CXCL8 and TNF-α which activates neutrophils leading to an upregulation of CD66b and increased production of ROS. Interestingly, increased levels of MPO-DNA complexes, major components of NETs, were found in the serum of DHF patients when compared with levels in dengue fever patients. Further, authors showed that DENV-primed neutrophils were prone to delobulation, an early feature of NET formation
[18]. Further supporting the relevance of NET formation and severe dengue manifestation, in-vivo studies have shown encapsulation of platelets expressing dengue viral antigen by the NETs. Authors proposed that neutropenia, a common clinical sign in dengue patients, may be beneficial to the host, as components of NETs may aggravate dengue immunopathogenesis
[20].
Overall, neutrophil recruitment during viral inoculation by Aedes aegypti mosquitoes seems to play a major role in early phases of infection. Further, patients with worse dengue disease outcomes present with increased neutrophil-associated transcripts along with upregulation of granulocyte activation markers, NET formation and ROS production. Similarities between proinflammatory cytokine plasma levels in both severe Dengue and Yellow fever patients indicate that the role of neutrophils should be investigated in yellow fever.
2.3. Implications in Pregnancy
In “Experimental studies of congenital malformations”, James G. Wilson compiled all known teratogenic agents at that time, defined as capable of causing abnormalities in embryonic or fetal development
[47]. Teratogenic agents were classified into several groups: drugs and chemicals; physical agents (e.g., temperature); growth and metabolic inhibitors; maternal nutritional deficiencies; endocrine imbalances; and infections (known as TORCH agents nowadays). Later on, in 1977, Wilson defined the signs of abnormal development as death, malformation, growth retardation and functional disorder, which includes neurological dysfunction and effects on behavior and cognition that may manifest later in life
[48]. The embryonic stage is the most sensitive period to teratogen action, including viral infection, and takes place from two to eight weeks post conception, a period when women may not be aware of pregnancy
[49]. In fact, infections during pregnancy are considered to be one of the major causes of maternal, fetal and neonatal mortality and morbidity
[50]. Following the events of the ZIKV epidemics in 2015, this flavivirus has been classified as a teratogen among TORCH agents. ZIKV infection led to thousands of Congenital Zika Syndrome (CZS) cases, mostly in Brazil
[51]. The literature reports that not only ZIKV, but also JEV, WNV and SLEV infections during pregnancy may lead to intrauterine-growth restriction (IUGR), congenital malformations and/or fetal demise, the ultimate manifestations of the action of teratogenic agents
[48].
During pregnancy, the maternal organism undergoes several adaptations to tolerate the presence of the fetus and also support its development until parturition. Those are immunological, physiological and hormonal adaptations taking place at the placenta and maternal–fetal interface
[52][53], but also systemically
[54]. Evidence suggests an increased susceptibility of pregnant women to viral infections due to aforementioned changes in the maternal immunological profile
[55], notably a shift in towards a predominantly humoral immune response rather than cytotoxic
[56]. Conversely, the number of circulatory PMN cells increases progressively throughout gestation, in association to increasing G-CSF plasma levels and peaking in the third trimester. During labor, neutrophil density is selectively greater in the lower uterine segment, presenting a rich source of inflammatory mediators such as eicosanoids, collagenase, elastase, IL-1β and TNF-α
[57][58][59]. Baseline neutrophil activation changes during pregnancy, characterized by increased expression of CD11b and higher responsiveness to stimuli, such as increased phagocytosis, degranulation, NETs release and higher production of ROS
[60][61].
In the female reproductive tract, PMN cells dispose of invading pathogens by phagocytosis or degranulation
[62]. However, their presence and activity are tightly regulated by steroidal hormone action on the epithelial cells from the female reproductive tract. The activation of estrogen receptor ESR1 in these cells downregulates epithelial factors required to initiate transepithelial migration, impairing the recruitment of neutrophils to vaginal tissues, but ensuring their integrity during pregnancy
[63]. This regulatory network prevents undesirable immune activation, which often lead to gestational disorders such as pre-eclampsia, placental insufficiency and fetal growth restriction
[64]. However, recurrent infections may disrupt this balance, enhancing placental constitutive expression of the neutrophil-chemoattractant CXCL8
[65], and leading to the accumulation of invading fetal and maternal neutrophils in amniotic fluid
[66]. In fact, high levels of chemokines CXCL10 and CXCL8 were found in the sera of preeclamptic mothers
[67], where CXCL8 activates circulatory neutrophils and induces the release of NETs in the intervillous space of preeclamptic placenta
[68]. Moreover, neutrophil abnormal activity is associated with severe pregnancy disturbances such as recurrent fetal loss, gestational diabetes mellitus, IUGR and preeclampsia
[68][69].
Although flaviviruses have been identified as human viral pathogens for more than a century
[70], the impacts of flavivirus infection on pregnancy outcomes are not well understood. Except for ZIKV, the literature of flaviviral infection during pregnancy is limited, often resumed on reports of transplacental infection. The mechanisms by which infection occurs and how it affects embryo or fetus development remain to be explored. For instance, DENV infection during pregnancy has been associated with increased risk of hemorrhagic fever and shock
[71]. DENV infection in early pregnancy was associated with fetal loss and probable transplacental transmission
[72][73], while perinatal infection may result in neonatal infection
[74][75]. Two reports describe YFV congenital infection: the first, dating from 1940, described two cases of fatal maternal infection mid-gestation, in which autopsy of one patient showed retro-placental hemorrhage, generalized steatosis in the fetus, extensive bleeding and leukocyte infiltration in fetal liver, lesions in the hepatic gland and hemorrhages in the intestinal tract
[76]; the second documented case describes the onset of yellow fever symptoms on a female patient three days before delivery, resulting in newborn fatal infection, in Brazil
[77].
Regarding encephalitogenic flaviviruses, the natural and experimental JEV infection in swine led to fetus mummification and stillbirths
[78][79]. The human transplacental infection with JEV was described for the first time in 1980, during an outbreak of Japanese encephalitis. The effects of JEV infection during pregnancy varied from parturition of apparently normal children to abortion, furthermore, it was possible to isolate JEV from brain, liver and placental tissues from an aborted fetus
[80]. A mouse model of intrauterine infection with SLEV showed severe neurological outcomes in fetuses, and the severity of the disease depended on the gestational day of infection
[81]. There is no data on maternal or fetal risks associated with SLEV congenital infection in humans
[82]. In 2002, the CDC published the first case report on intrauterine WNV infection, the 2-day old infant presented positive serology for WNV, chorioretinal scarring and severe brain abnormalities
[83][84]. An anomaly rate of 10.6% was reported in a clinical study of pregnant women infected with WNV in the USA, which is almost twice the rate of 5.5% for the general population and included four cases of miscarriage and four preterm deliveries. Of 55 infants born at term, most did not present anti-WNV IgM in cord serum, but some individuals developed a degree of malformation such as abnormal growth, aortic coarctation, glycogen storage disease type 1, cleft palate, Down syndrome, microcephaly, polydactyly or lissencephaly
[85]. Moreover, WNV and Powassan virus (POWV, a tick-borne flavivirus) are capable of infecting and damaging human placental explants or pregnant mice placentas
[86]. Whether these outcomes are caused by flaviviral infection
per se or by dysregulation of immune response remains to be explored, along with possible molecular mechanisms involved.
The ZIKV epidemic in 2015 brought a significant increase in cases of microcephaly and congenital malformations in Brazil, and prompted the establishment of in-vitro and in-vivo models of infection to better understand ZIKV biology and pathogenesis
[87]. Zika was shown to be a neurotropic flavivirus, capable of infecting not only neuronal cells but also trophoblasts in placenta and the developing embryo, which ultimately causes the Congenital Zika Syndrome (CZS). The most common clinical manifestations of CZS include microcephaly, ventriculomegaly, intracranial calcification, ocular abnormalities and hearing loss
[88][89][90]. Pregnant women with symptomatic ZIKV infection showed higher viral load in sera than nonpregnant women
[91]. In addition to other proinflammatory chemokines, Camacho-Zavala and colleagues found high plasma levels of CXCL8, which correlate directly to neutrophil activation
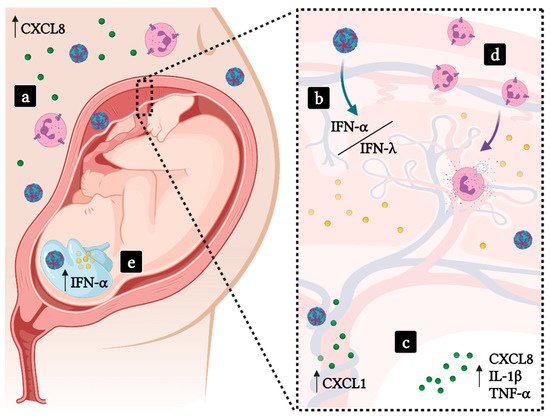
[91] (
Figure 3). Moreover, the amniotic fluid from ZIKV-positive pregnant women whose children presented microcephaly revealed exceedingly high levels of cytokines and growth factors: IL-15, CCL11, CXCL10, G-CSF, IL-10, IL-1β, TNF-α, CXCL8, CCL2 and CCL5
[92]. These data corroborate the proinflammatory profile found in pregnant Zika patients and are suggestive of neutrophil involvement, as G-CSF, CXCL8, IL-1β and TNF-α are involved in neutrophil activation and effector functions. Finally, cerebrospinal fluid from infants with ZIKV-induced microcephaly showed a maintenance of the inflammatory environment by the presence of IFN-α, CXCL10 and CXCL9 after birth
[93].
Figure 3. ZIKV congenital infection and the probable participation of neutrophils in CZS. (a) High levels of CXCL8 are found in the serum of pregnant women infected with ZIKV (blue/red viral particles). (b) ZIKV is able to trespass the placental barrier, the persistence of the infection leads to an imbalance on Type-I and Type-III IFN responses, which may lead to tissue damage. (c) Amniotic fluid from CZS pregnancies present high levels of neutrophil-chemoattractants and proinflammatory cytokines and ZIKV-infected human umbilical vein endothelial cells are sources of CXCL1. (d) Recruited neutrophils respond to pathogenic activation of Type-I IFN responses in patients. (e) ZIKV may infect fetal tissues, inducing a proinflammatory environment that promotes neuronal damage and the onset of CZS. Green dots—CXC chemokines; Yellow dots—IFNs.
Transcriptome profile of human umbilical vein endothelial cells (HUVEC) infected in vitro with ZIKV revealed upregulation of IL-15, CCL5, HGF, LIF, M-CSF, CXCL1 and CXCL12 24 h post infection. Cells infected with the Puerto Rican strain, a representative of Asian ZIKV strains, showed increased levels of IL-1β, IL-10, CCL5, G-CSF, CSF, CXCL1 and CXCL12 cytokines and reduction of IL-15, IL-16, HGH, PDGFbb and CXCL9 cytokines. Inoculation of the African strain in HUVEC cell cultures resulted in a higher expression of CCL2, CCL5, bFGF, G-CSF, LIF, M-CSF, CXCL1, and CXCL12 and a decrease in IL-15, HGH and CXCL11
[94]. Recurrent expression of CXCL1 and CXCL12 suggests that ZIKV infection of umbilical endothelial cells may lead to neutrophil recruitment to the umbilical cord.
Placentas infected with ZIKV develop a type-III IFN response in the decidua. Usually, IFN-λ has been shown to be protective against infections in mucosal surfaces, inducing antiviral responses with minimal damage to tissues. In the context of the placenta, IFN-λ is also tissue-protective, sustaining the placental role as a barrier and by counteracting infection through induction of ISGs. Human trophoblast cells isolated from full-term placentas were refractory to ZIKV infection due to IFN-λ1 expression
[95]. Neutrophils express high levels of the heterodimeric IFN-λ receptor IFNLR1/IL10RB
[96], resulting in a repressed state of neutrophils’ effector functions induced by type-III IFN signaling, exemplified by reduction in neutrophil infiltration, degranulation, ROS production and NETs release
[97]. In the early stages of ZIKV placental infection, typical flavivirus antagonism of IFN responses result in downregulation of IFN-λ2, leading to an aberrant neutrophil response and CZS
[98]. CZS is later accompanied by a strong type-I IFN response, increased expression of IFIT5 and decrease of ISG15 mRNA expression levels
[99]. Studies using mouse models of ZIKV infection show that fetal and placental type-I IFN responses play a major role in promoting placental damage and fetal demise in ZIKV congenital infection
[99][100]. The activation of a robust type-I IFN response may lead to tissue damage through induction of chemokine expression and recruitment of neutrophils, proinflammatory monocytes and T lymphocytes
[101]. High levels of IFN-α early in pregnancy may cause angiogenic imbalance in the placenta, associated with higher risks for preeclampsia
[102]. Hence, the proinflammatory environment promoted by ZIKV infection during pregnancy indicates that neutrophils may be recruited to infected tissues and that neutrophil functions may be dysregulated.
 +1 point
+1 point