1000/1000
Hot
Most Recent
 +1 point
+1 point
Neurotransmission is the process by which a signal is conveyed between neurons via endogenous signaling molecules called neurotransmitters. Neurotransmitters released from the axon terminal of one neuron cross the synaptic cleft and bind to receptors on the dendrites of another neuron, which are then converted into electrical signals. Synapse, the junction between neurons, has a tripartite structure that consists of presynaptic and postsynaptic nerve terminals along with the intimate association of glial cells.
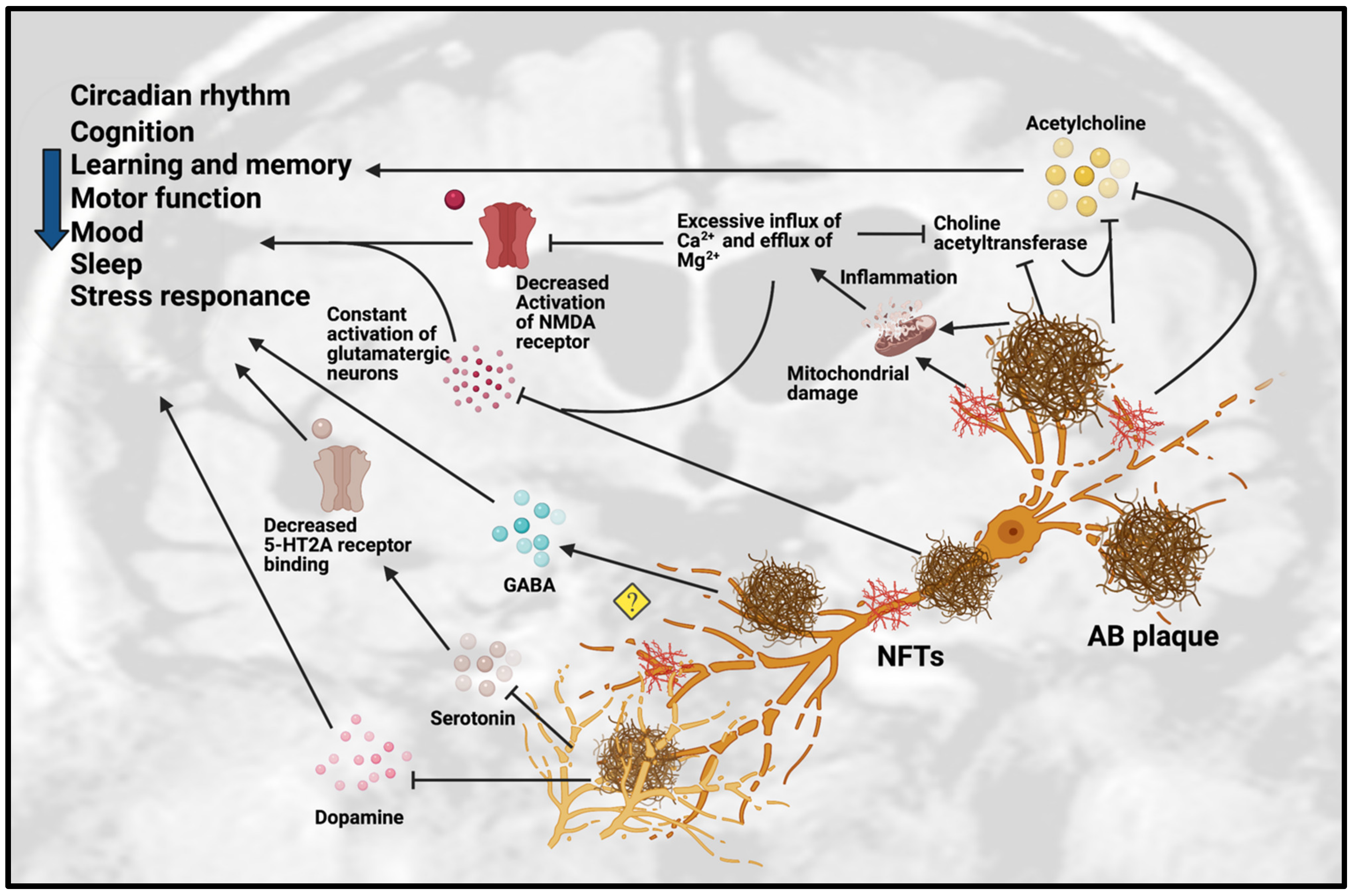 Figure 1. Dysfunction of neurotransmission in AD. Accumulation of Aβ plaques and NFT in AD cause impairment of the circadian rhythm, cognition, learning, memory, motor function, mood, sleep and stress response. These pathologies are toxic to neurotransmission systems, affecting cholinergic, glutamatergic, serotonergic and dopaminergic systems. Amyloid-beta plaques and NFT can inhibit the release of ACh and choline acetyltransferase, an enzyme that regulates ACh synthesis, which reinforces the inhibition effect of ACh. Amyloid-beta plaques and NFT can cause mitochondrial damage in glutamatergic neurons. The mitochondrial damage leads to inflammation due to excessive influx of Ca2+ and excessive efflux of Mg2+ that affect the activation of glutamatergic neurons and decreases the activation of NMDA receptor. The excessive influx of Ca2+ in glutamatergic neurons leads to inhibition of choline acetyltransferase and further inhibits the synthesis of ACh. However, the detailed mechanisms are not yet understood, as some studies showed the upregulation of GABA in certain regions but downregulation of GABA in other regions. Amyloid-beta plaques also disrupt the homeostatsis of serotonin (5-HT) by inhibiting the binding of serotonin receptor (5-HT2A) and disrupting the dopaminergic system. Abbreviations: Aβ, Amyloid-beta; NFT, neurofibrillary tangle.
Figure 1. Dysfunction of neurotransmission in AD. Accumulation of Aβ plaques and NFT in AD cause impairment of the circadian rhythm, cognition, learning, memory, motor function, mood, sleep and stress response. These pathologies are toxic to neurotransmission systems, affecting cholinergic, glutamatergic, serotonergic and dopaminergic systems. Amyloid-beta plaques and NFT can inhibit the release of ACh and choline acetyltransferase, an enzyme that regulates ACh synthesis, which reinforces the inhibition effect of ACh. Amyloid-beta plaques and NFT can cause mitochondrial damage in glutamatergic neurons. The mitochondrial damage leads to inflammation due to excessive influx of Ca2+ and excessive efflux of Mg2+ that affect the activation of glutamatergic neurons and decreases the activation of NMDA receptor. The excessive influx of Ca2+ in glutamatergic neurons leads to inhibition of choline acetyltransferase and further inhibits the synthesis of ACh. However, the detailed mechanisms are not yet understood, as some studies showed the upregulation of GABA in certain regions but downregulation of GABA in other regions. Amyloid-beta plaques also disrupt the homeostatsis of serotonin (5-HT) by inhibiting the binding of serotonin receptor (5-HT2A) and disrupting the dopaminergic system. Abbreviations: Aβ, Amyloid-beta; NFT, neurofibrillary tangle.| Animal Model. | Gender | Age | Pathology Involved | Neurotransmission Dysfunction | Behavioral Effects | References |
|---|---|---|---|---|---|---|
| APPswe/PS1dE9 mice | N/A | 4–18 months old | Degeneration and loss of forebrain 5-HT and NA axons after Aβ deposits | Monoaminergic neurodegeneration | Anxiety-related behaviors in 18 months | [2] |
| Swiss mice treated with AβO | N/A | 3 months old | Development of Aβ plaques | AβO disrupts 5-HT homeostasis | Depressive-like behavior | [3] |
| APPswe/PS1dE9 mice | Male | 4, 8, 11 months old | Progressive accumulation of Aβ protein. | Significant decrease in 5-HT2A receptor binding | Memory impairment | [4] |
| 5xFAD mice | Male | 6 months old | Significant decrease of both TH+ and TH- cells in DA-producing areas | SN-VTA networks are enhanced to the synchronization of neuronal firing activity in DA-producing nuclei |
|
[5] |
| Tg2576 mice | Male | 2 and 6 months old | Degeneration of VTA DAergic neurons | Reduced noradrenergic transmission in dorsal subiculum | Age-related impairment of memory and non-cognitive functions | [6] |
| Tg2576 mice | N/A | 4–6 and 9–11 months old | Aβ were prominent in 20-month-old mice | Reduced ACh release from hippocampus in 9- to 11-month-old mice | Memory impairment present in 9- to 11-month-old mice | [7] |
| APP/PS1 mice | N/A | 3 and 7 months old | Aβ plaques deposition after cholinergic degeneration |
|
|
[8] |
| APP/PS1 and 5xFAD mice | N/A | 8 and 13 months old | Aβ plaques deposition and reactive astrocytes | Aberrant increase in GABA release from reactive astrocytes | Impaired learning and memory | [9] |
| AβPP/PS mice | Male | 2–4 months old | Abnormal glutamate release precedes cognitive decline | Significantly increased potassium-evoked glutamate release in CA1 | Cognitive decline | [10] |
| AβPPswe-PS1dE9 mice | N/A | 6 months old | Deposition of Aβ plaques |
|
Impairment of cognitive function and memory | [11] |
| TgAPP23 mice | Male and female | 24 months old | Deposition of Aβ plaques and cholinergic degeneration |
|
N/A | [12] |
| PS2APP mice | Female | 20 or 24 months old | Deposition of Aβ plaques | Significant reduction of glutamate level in frontal cortex | N/A | [13] |
| TgAPP23 mice | N/A | 7–8 months old | Dysfunction of cholinergic and monoaminergic systems |
|
N/A | [14] |
| PDAPP mice | Male and female | 4–6 months old | Deposition of Aβ plaques | Reduced basal and evoked ACh release from hippocampus | Hyper-locomotor function | [15] |
| 3xTg-AD mice | Male and female | 2–4, 13–15 and 18–20 months old | Aβ plaques deposition with cholinergic degeneration and alteration of neurotrophic factors |
|
N/A | [16] |
| hAPP-J20 mice | N/A | 6 months old | Altered synaptic plasticity and cognitive function | Significantly decreased phospho GluN2B levels and hippocampal LTP | Impaired learning and memory | [17] |
| TgCRND8 mice | N/A | 2 and 7 months old | Aβ plaques deposition, oxidative stress, reactive glial cells and neurodegeneration | Reduced ChAT-positive neurons and ACh levels. | Cognitive impairment | [18] |
| PS2APP mice | Male | 5, 9, 13 and 17 months old | Deposition of Aβ plaques | Significant loss of mGlu2 receptors in entorhinal cortex and lacunosum moleculare regions | N/A | [19] |
| PS2APP mice | Male | 3–4 months old | Altered synaptic plasticity | Aberrant GluN2B-NMDAR function | N/A | [20] |
| PDAPP mice | Male | 2, 4, 12 and 24 months old | Aβ plaques deposition with cholinergic degeneration |
|
N/A | [21] |
| 3xTg-AD mice | N/A | 9–23 months old | Deposition of Aβ plaques | Reduced ChAT and AChE-positive neurons | N/A | [22] |
| TgCRND8 mice | Male | 3 months old | Deposition of Aβ plaques and neuronal degeneration |
|
Cognitive impairment | [23] |
| TgCRND8 mice | Male and female | 2–3 and 12–13 months old | Deposition of Aβ plaques |
|
N/A | [24] |
| TgCRND8 mice | Male | 3 months old | Dysfunction of dopaminergic system |
|
Cognitive impairment | [25] |
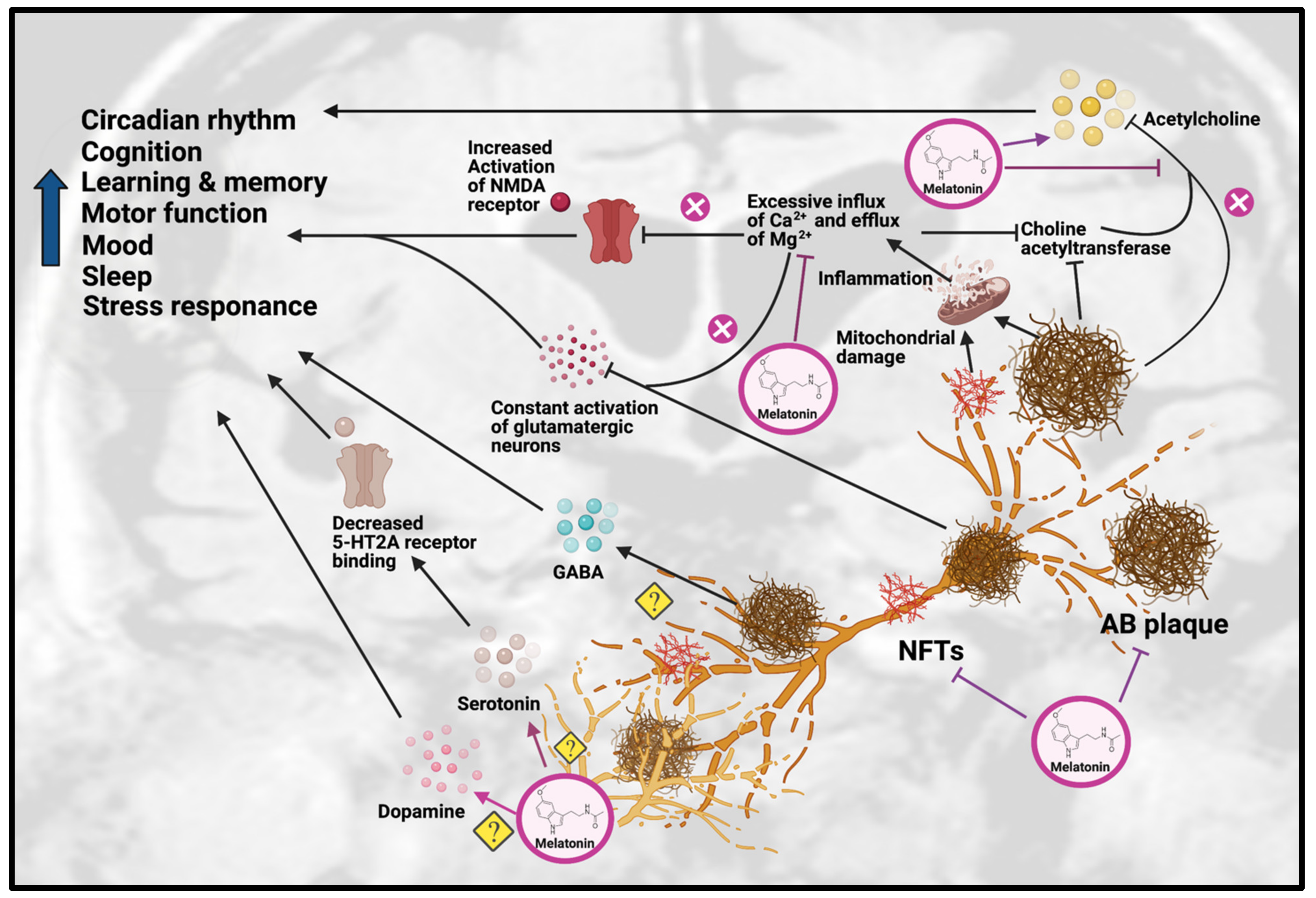 Figure 2. Effects of Melatonin treatment on dysfunction of neurotransmission in AD. Melatonin can ameliorate the formation of Aβ plaques and NFT, as well as improve the impairments due to these AD hallmarks, including disrupted circadian rhythm, cognition, learning, memory, motor function, mood, sleep and stress response. Melatonin treatment can have beneficial effects on serotonergic and dopaminergic systems, but the exact mechanisms have yet to be determined. Melatonin can also have beneficial effects on the cholinergic system by increasing acetylcholine release and reducing inflammation caused by excessive influx of Ca2+ and excessive efflux of Mg2+, thereby inhibiting choline acetyltransferase. Abbreviations: Aβ, Amyloid-beta; NFT, neurofibrillary tangle.
Figure 2. Effects of Melatonin treatment on dysfunction of neurotransmission in AD. Melatonin can ameliorate the formation of Aβ plaques and NFT, as well as improve the impairments due to these AD hallmarks, including disrupted circadian rhythm, cognition, learning, memory, motor function, mood, sleep and stress response. Melatonin treatment can have beneficial effects on serotonergic and dopaminergic systems, but the exact mechanisms have yet to be determined. Melatonin can also have beneficial effects on the cholinergic system by increasing acetylcholine release and reducing inflammation caused by excessive influx of Ca2+ and excessive efflux of Mg2+, thereby inhibiting choline acetyltransferase. Abbreviations: Aβ, Amyloid-beta; NFT, neurofibrillary tangle.




