1000/1000
Hot
Most Recent
 +1 point
+1 point
Apoptosis is a process of programmed cell death which has an important role in tissue and organ homeostasis and in the control of organism development. Two main signaling pathways are involved in regulation of apoptosis: the mitochondrial dependent intrinsic pathway and death receptor dependent extrinsic pathway. The receptors are members of the tumor necrosis factor receptor superfamily (TNFRSF).The extrinsic apoptotic pathway triggers apoptosis by binding of ligands to death receptors, which leads to formation of a death-inducing signaling complex (DISC) and in consequence, caspase activation. This pathway and its subpathways and regulatory factors and linkages to the other cellular signaling events are briefly presented.
The TNFRSF consists of TNF receptor-associated factor (TRAF)-interacting receptors, decoy receptors (DcRs) and death receptors (DRs). The DRs can further be classified according to the molecular mechanisms of apoptosis and necroptosis activation: TNF receptor superfamily member 6 (Fas), TNF-related apoptosis-inducing ligand receptor 1 (TRAILR1) and TNF-related apoptosis-inducing ligand receptor 2 (TRAILR2) belong to the first subgroup of the DRs, which on the cytosolic plasma membrane surface, directly interact with an adapter protein, Fas-associated protein with death domain (FADD) and tumor necrosis factor α (TNF-α) receptor 1 (TNFR1/p55), which belongs to the second subgroup of DRs that act directly with TNFR1-associated protein with death domain (TRADD) [1]. Schemes of mentioned above pathways are shown in Figure 1.
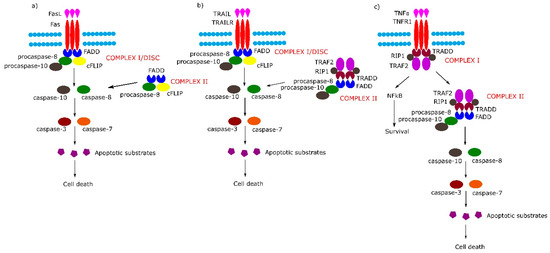
Figure 1. Extrinsic apoptotic signaling pathway. (a,b) Tumor necrosis factor (TNF) receptor superfamily member 6/tumor necrosis factor-related apoptosis-inducing (Fas/TRAIL) mediated apoptotic pathway leads to the formation of Complex I/death-inducing signaling complex (DISC) which consists of Fas-associated protein with death domain (FADD), procaspase 8/10 and cellular FLICE (FADD-like IL-1β-converting enzyme) inhibitory proteins (cFLIP). (c) Binding of tumor necrosis factor α (TNFα) to TNF-α receptor 1 (TNFR1) receptor causes the formation of Complex I (TNFR1-associated protein with death domain (TRADD), TNFR1, TNF receptor-associated factor 2 (TRAF2), TNFα-related receptor interacting protein (RIP), which in the next stage either activates the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) pathway or transforms into Complex II (TRADD, TNFR1, TRAF2, RIP, FADD, procaspase-8, procaspase-10).
The DRs are characterized by an extracellular N-terminal domain with cysteine-rich domains (CRDs), a membrane-spanning region and a C-terminal, an intracellular domain which harbors a death domain (DD). While DRs share the CRDs with all members of the TNFRSF, the DD is only found in DRs. The CRDs contain the N-terminal pre-ligand assembly domain (PLAD) which is involved in receptor–receptor interactions and is involved in binding ligands of the TNF superfamily (TNFSF) [2][3]. The PLAD domains’ interactions cause spontaneous receptor dimerization and/or trimerization. In the case of CD95/Fas and TRAILR death receptors, PLAD–PLAD interaction has a high affinity, in contrast to the TNF1 receptor [4].
Death ligands exist in the cell membrane as trimeric type II transmembrane proteins belonging to the TNFSF. They contain a conserved C-terminal TNF homology domain (THD), an N-terminal intracellular proline-rich domain (PRD) and a stalk region. THD binds to CRDs of the TNFRSF. Membrane-bound death ligands can be processed in the stalk region by metalloproteases, which leads to the release of soluble molecules. The soluble death ligands also own THD and, in consequence, have the capacity to react with TNFRSF receptors [2].
According to the sequential model of TNFRSF receptor activation, a single TNFRSF receptor interacts with a TNFSF ligand trimer and forms a cell surface-bound TNFRSF receptor-TNFSF ligand3 complex. Next, in two subsequent stages, two additional monomeric TNFRSF receptors are added to create an active TNFSF ligand3–TNFRSF receptor3 cluster which is necessary for induction of death-inducing signaling complexes [4].
The binding of Fas with tumor necrosis factor ligand superfamily member 6 (FasL) leads to formation of the DISC, also termed complex I, consisting of FADD, procaspase-8, procaspase-10 and cellular FLICE (FADD-like IL-1β-converting enzyme) inhibitory proteins (c-FLIPs). The formation of DISC leads to activation of caspase-8, caspase-10 and in consequence to cleavage and activation of effector caspase-3 and caspase-7. Additionally, in some cell lines, a second cytosolic complex is formed upon ligand stimulation. This complex, called complex II, composed of FADD, procaspase-8 and c-FLIPs, might amplify caspase activation by processing caspase-3 (Figure 1a) [5].
Upon tumor necrosis factor-related apoptosis-inducing (TRAIL) stimulation, the TRAIL receptor forms membrane-associated DISC/complex I in a similar way to the Fas receptor and secondary complex, similarly to above-mentioned TNFR1 complex II (Figure 1b) [5].
Signaling through TNFR1 leads to the formation of complex I and then complex II. Complex I consists of TRADD, TNFR1, TNF receptor-associated factor 2 (TRAF2) and tumor necrosis factor α (TNFα)-related receptor interacting protein (RIP). Complex I assembles rapidly following TNF-α stimulation, and activates the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) signal transduction pathway, which activates survival signals. In the next step, complex I dissociates from the death receptor and binds FADD and procaspase-8 (complex II), the activation of which leads to an induction of apoptosis (Figure 1c) (reviewed in [5]).
It is worth noting that the activation of procaspase-8 at DISC is driven by death effector domain (DED) “chains”, which include procaspase-8, procaspase-10 and c-FLIP. The number of procaspase-8 molecules in DED chains is 10 times higher than the number of procaspase-10 and c-FLIP molecules. The local increase of procaspase-8 concentration in DISC causes dimerization of procaspase-8. Then, procaspase-8 dimers (also called p55/p53) are converted by autocatalytic two-step transprocessing into mature heterotetrameric caspase-8 (p182/p102) while the DISC-bound caspase-8 prodomain can be replaced by a new procaspase-8 molecule. The termination of the DED chain’s elongation depends on its stability and association/dissociation rates of procaspase-8 to the chain (Figure 2) [1][6][7].
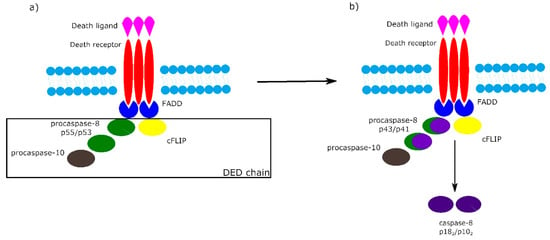
Figure 2. Death effector domain (DED) chains and procaspase-8 transprocessing. a) Activation of procaspase-8 begins with DED chains, which consist of procaspase-8 p55/p53, procaspase-10 and c-FLIP. b) Procaspase-8 homodimers are converted into active caspase-8 heterotetramers p182/p102 via the intermediate procaspase-8 p43/p41.
Among the most important regulators of the receptor-dependent apoptosis pathway are c-FLIP isoforms, which are major antiapoptotic proteins. Three isoforms can be distinguished as: short c-FLIPS, Raji c-FLIPR and long c-FLIPL. All c-FLIP isoforms possess two N-terminal DED domains. c-FLIPS and c-FLIPR are truncated version of procaspase-8, whereas c-FLIPL retains a full length procaspase-8 chain in which the catalytic cysteine residue within the active site is missing and thus has no proteolytic activity [7]. The isoforms of c-FLIP can heterodimerize with procaspase-8 via a co-operative and hierarchical binding mechanism. The result of c-FLIPS or c-FLIPR homo- or heterodimerization with procaspase-8 is prevention of DED-mediated caspase-8 oligomerization and inhibition of its activation. The ratio of c-FLIPL to procaspase-8 in c-FLIPL-procaspase-8 heterodimers determines whether procaspase-8 will be activated or inhibited. At physiologically-low levels, c-FLIPL-procaspase-8 heterodimer activates procaspase-8. In contrast, high levels of c-FLIPL block procaspase-8 oligomerization, which results in inhibition of caspase cascade activation (Figure 3) [7][8].
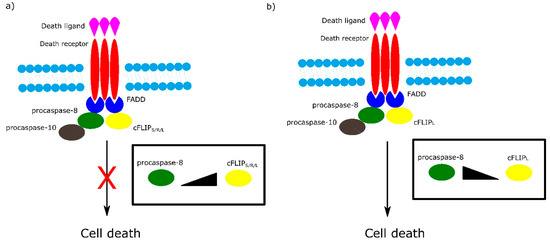
Figure 3. Role of different isoforms of cFLIP in extrinsic apoptotic pathway. The ratio of cFLIP to procaspase-8 in cFLIP-procaspases-8 heterodimer determines cell fate. a) High levels of cFLIPS/R/L block formation of procaspase-8 oligomer and in consequence inhibit cell death. b) At low level of cFLIPL , cFLIPL - procaspase-8 heterodimer activates cell death.
Other regulators of apoptosis are inhibitors of apoptosis (IAPs). The mammalian IAPs, X-linked IAP (XIAP), cellular inhibitor of apoptosis protein 1 (cIAP1) and cellular inhibitor of apoptosis protein 2 (cIAP2) contain three baculovirus IAP repeat (BIR) domains which mediate protein–protein interactions, UB-associated domain (UBA), which is responsible for interaction with ubiquitylated proteins, and the “really interesting new gene” (RING) domain conferring E3-ubiquitin ligase activity. Additionally, cIAP1 and cIAP2 have the caspase recruitment domain (CARD), which has the ability to inhibit their E3 ligase activity [9]. The BIR1 domain of XIAP binds the TGFβ-activated kinase 1 (TAK1)-binding protein 1 (TAB1) [10], whereas cIAPs’ BIR1 domain binds TRAF1 and TRAF2 [11]. In turn, the XIAP BIR2 domain is involved in inhibition of effector caspase-3 and caspase-7, while BIR3 binds to caspase-9 and prevents dimerization of this enzyme [12]. cIAP1 and cIAP2 can bind caspases through BIR2 and BIR3 domains, but do not inhibit them [13]. XIAP is able to induce ubiquitination of active caspases at the K48 residue, causing their (proteasomal) degradation. Additionally, XIAP is involved in covalent tagging of caspase-7 with ubiquitin-like protein NEDD8 leading to inactivation of this caspase [14]. cIAP1 and cIAP2 are able to ubiquinate caspase-3 and caspase-7 [15][16] but only cIAP1 targets them for proteasomal degradation [15].
The linear ubiquitin chain assembly complex (LUBAC) is another regulator of cell death. LUBAC is the only known E3 ligase complex for linear ubiquitination, which consists of ring finger protein 31 (RNF31, also known as HOIP), HOIL-1 and Shank-associated RH domain-interacting protein (sharpin) [17]. Recent studies showed that sharpin has a regulatory function in NF-κB and apoptotic signaling pathways. Absence of sharpin attenuates TNFα-mediated NF-κB activation [18][19][20]. Cleavage of RNF31 by caspase-3 and caspase-6 suppresses activity of LUBAC in the NF-κB signaling pathway. This process weakens the inhibitory role of NF-κB in death signaling, leading to the sensitization of cells to cell death signals [21]. On the other hand, another study showed that RNF31 limits caspase-8 activity in complex I and complex II in TRAIL signaling, which causes inhibition of apoptosis [22][23].





