Carcinogenesis is a multi-step process that refers to transformation of a normal cell into a tumoral neoplastic cell. The mechanisms that promote tumor initiation, promotion and progression are varied, complex and remain to be understood. Studies have highlighted the involvement of oncogenic mutations, genomic instability and epigenetic alterations as well as metabolic reprogramming, in different processes of oncogenesis. However, the underlying mechanisms still have to be clarified. Mitochondria are central organelles at the crossroad of various energetic metabolisms. In addition to their pivotal roles in bioenergetic metabolism, they control redox homeostasis, biosynthesis of macromolecules and apoptotic signals, all of which are linked to carcinogenesis.
1. Introduction
Carcinogenesis is the process by which a normal cell evolves until it becomes a cancerous cell. The etiology of carcinogenesis is complex, multifactorial and can involve cellular, molecular, genetic, epigenetic and environmental alterations
[1]. Among the six hallmarks of cancer that were updated by Hanahan and Weinberg in 2011, the deregulation of cellular energetics stands as a major mechanism supporting neoplastic transformation
[2]. In cancer, metabolisms of amino acids, lipids, nucleic acids, glutamine and glucose can be altered to promote cell proliferation and survival
[3][4]. Rewiring cell metabolism is one way by which cancer cells survive to gene alterations, low nutrients availability, hypoxic environment and increased stiffness of surrounding tissues. Cancer cells thus reprogram their metabolism to gain energetic metabolites that fuel cancer initiation and maintenance
[5].
Mitochondria are major players in metabolic reprogramming of cancer cells. These organelles, often qualified as “the powerhouse of the cell”, have been the subject of intense research over the past 50 years because of their pleiotropic functions. Long been considered as the center of cellular energy production, mitochondria are more than the energetic factories of the cell
[6]. They constitute an integrative hub controlling ATP generation, amino-acid synthesis, ROS production, redox balance, calcic signaling and apoptotic pathways
[7][8]. Mitochondria also represent “stress sensors” that coordinate metabolic adaptation of cells to their microenvironment.
2. Mutations in Genes Involved in Mitochondrial Metabolism Drive Carcinogenesis Initiation
The causal link between alteration of mitochondrial metabolism and initiation of carcinogenesis is illustrated by the occurrence of mutations in mtDNA and nuclear-encoded mitochondrial genes that alter oxidative respiration and promote oxidative stress and epigenetic processes.
2.1. Mutations and Decreased Copy Number of mtDNA
Mutations in mtDNA and decreased mtDNA copy number are frequently found in cancer cells and are believed to drive carcinogenesis
[9]. Driving effects of mtDNA mutations in carcinogenesis initiation have been clearly established in cancer cell lines depleted for mtDNA (ρ0 cells), where introducing an mtDNA mutation was found to promote cancer growth and ROS production
[10]. mtDNA is more often mutated than nuclear DNA, probably due to lack of histone protection, limited capacity of DNA repair and proximity with the respiratory chain which is the major producer of ROS
[11]. Indeed, mitochondrial ROS induce oxidative damage to lipids and proteins and mutagenize mtDNA, thereby coupling respiratory chain deficiency and carcinogenesis
[12]. Mutations of mitochondrial genes encoding NADH dehydrogenase (component of complex I), cytochrome B (complex III), COX I (complex IV) and ATP synthase (complex V) have been associated with a deficit of respiratory function, together with lactate accumulation and ROS overproduction which further stimulates oncogenic signaling pathways
[10][13][14][15]. mtDNA mutations in cancer have been extensively reviewed and listed by Hertweck and Dasgupta in 2017
[16]. Elevated percentage of mtDNA mutations is often correlated with high degree of bioenergetic defects. Given that there are hundreds of copies of mtDNA in each cell, mutations can affect all mtDNA molecules (homoplasmy) or a variable proportion of mtDNA molecules (heteroplasmy) which leads to different disease phenotypes
[17]. For instance, homoplasmic mutations of ND5 protein (complex I) inhibit tumor growth whereas heteroplasmic ND5 mutations promote tumorigenesis. Indeed, heteroplasmic mtDNA mutations moderately alter mitochondrial functions and promote ROS-dependent cell proliferation whereas homoplasmic mutations induce severe mitochondrial damages with lethal consequences for cancer cells
[14]. In addition to mutations in the mtDNA coding regions, highly frequent somatic mutations occur in a non-coding region of mtDNA called the D-loop, that constitutes a mutational “hotspot”
[16]. The D-loop region controls mtDNA replication and transcription. Mutations in this region decrease mtDNA copy number and thus alter expression of mitochondrial genes, with deleterious consequences for mitochondrial integrity and a promoting effect in carcinogenesis
[9]. Importantly, mtDNA depletion is also correlated with both hyper- and hypomethylation of nuclear genome in a not-yet elucidated epigenetic mechanism
[18]. Mitochondrial-driven regulation of the nuclear genome identified with mtDNA mutations highlights a bidirectional interaction between mitochondria and the nucleus.
2.2. Mutations in Nuclear-Encoded Mitochondrial Genes
Mutations in mitochondrial enzymes encoded by nuclear DNA also contribute to carcinogenesis in various ways. They compromise the mitochondrial TCA cycle and oxidative respiration and induce ROS production and HIF-1α-dependent pseudohypoxia. Mutations in isocitrate dehydrogenase (IDH), succinate dehydrogenase (SDH) and fumarate hydratase (FH), three important enzymes of the TCA cycle, induce the accumulation of metabolic intermediates, namely 2-hydroxyglutarate (2-HG), succinate and fumarate, that are called mitochondrial oncometabolites. These three oncometabolites are significantly increased in tumor cells compared to normal cells
[19][20]. Accumulation of oncometabolites promotes initiation of carcinogenesis by altering mitochondrial-dependent biosynthesis pathways, redox balance and regulating nuclear genome epigenetic processes.
2.2.1. Bioenergetic Metabolism Alteration
IDH, SDH and FH mutations all induce a decrease in mitochondrial respiration because of a blockade of the TCA cycle and OXPHOS. As a result, these mutations are associated with an upregulation of glycolysis and lactate production
[21][22]. Oncometabolites promote glycolysis mainly by inhibiting pyruvate dehydrogenase (PDH), a key enzyme that converts mitochondrial pyruvate to acetyl-coA, the first intermediate that enters the TCA cycle. PDH inhibition interrupts the TCA cycle but also prevents HIF-1 degradation, resulting in HIF-1 accumulation that induces expression of genes involved in glycolysis
[23][24]. In response to mitochondrial bioenergetic decrease, compensatory pathways can be activated, such as glutaminolysis in IDH-1 mutated glioma, to maintain metabolic homeostasis
[25].
2.2.2. Oxidative Stress Promotion
Oncometabolites were also shown to disrupt redox homeostasis, which depends on the balance between production of ROS (superoxide anions, H
2O
2, hydroxyl radicals) and antioxidant pathways. In particular, SDH being both a TCA cycle enzyme and a component of ETC complex II, its mutations were shown to block electron transport across ETC complexes, leading to overproduction of superoxide anions
[26]. Oncometabolites thus promote carcinogenesis in two ways. They either alter ROS scavenging processes by inhibiting Nrf2 transcription factor, depleting intracellular NADPH and/or compromising gluthatione (GSH) disulfide reduction—all antioxidant systems that protect cells from oxidative stress
[27], or they promote ROS generation through alteration of ETC complex II activity
[28][29][30]. Interestingly, ROS inhibits PDH, suggesting that oncometabolites may induce a pseudo-hypoxic environment through ROS production. Thus, mutations in mitochondrial enzymes induce a tumorigenic hypoxia-like state mediated by HIF-1 stabilization and accumulation of ROS that promotes tumor growth
[31].
2.2.3. Epigenetic Regulation
Another effect of oncometabolites is the induction of carcinogenesis by epigenetic regulation of oncogenes and DNA repair enzymes. Accumulation of 2-HG, succinate or fumarate inhibits α-ketoglutarate-dependent dioxygenases involved in histone and DNA demethylations. They induce histone hypermethylation, resulting in homologous recombination (HR) DNA repair defects through KDM4A and KDM4B inhibition. These two lysine demethylases are necessary for efficient DNA repair and their inhibition leads to genomic instability
[32]. Inhibition of demethylases and dioxygenases (TET family) by oncometabolites also mediates epigenetic control of genes implicated in glycolysis
[33][34][35]. The implication of mitochondrial functions in the initiation of carcinogenesis are summarized in
Figure 1.
Figure 1. Theoretical basis for the use of metformin in PE prevention. AMPK—5’ adenosine monophosphate-activated protein kinase, VEGF—vascular endothelial growth factor, PlGF—placental growth factor, TGF β1—transforming growth factor-β1, sEnd—soluble endoglin, sFlt-1—fms-like tyrosine kinase-1, HTF1 α—hypoxia-inducible factor 1α, ICAM1—intracellular cell adhesion molecule 1, VICAM1—vascular cell adhesion molecule 1, NO—nitric oxide, ROS—reactive oxygen species, MMPs—matrix metalloproteinases, MMP-2—matrix metalloproteinase-2, MMP-9—matrix metalloproteinase-9, ERS—endoplasmic reticulum stress, EPCs—endothelial progenitor cells, SIRT1—sirtuin1.
3. Mitochondria as Promising Targets in Cancer Therapies
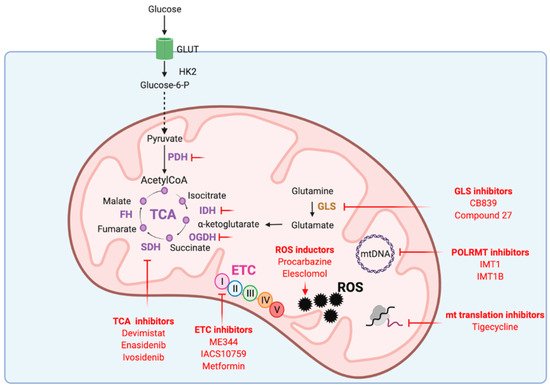
Given its key roles in carcinogenesis and tumor maintenance, mitochondria have emerged as interesting therapeutic targets. Addiction of cancer cells to nutrients for their growth have led to consider mitochondrial metabolism as the Achilles’s heel of tumors. Targeting mitochondrial metabolism has thus been widely investigated as potential cancer therapy. Disturbing mitochondrial metabolism and redox balance with pharmacological inhibitors (Figure 2) already gave promising results that are being evaluated in ongoing clinical trials.
Figure 2. Mitochondrial functions targeted in cancer therapy. ETC: electron transport chain, FH: fumarate hydratase, GLS: glutaminase, GLUT: glucose transporter, HK2: hexokinase 2, IDH: isocitrate dehydrogenase, mtDNA: mitochondrial DNA, OGDH: α-ketoglutarate dehydrogenase, PDH: pyruvate dehydrogenase, POLRMT: mitochondrial RNA polymerase, ROS: reactive oxygen species, SDH: succinate dehydrogenase, TCA: tricarboxylic acid cycle.
3.1. Targeting mtDNA Transcription and Translation
As mentioned earlier, mtDNA encodes proteins implicated in mitochondrial respiration. Inhibitors of mtDNA transcription that target the mitochondrial RNA polymerase POLRMT were recently developed
[36]. These inhibitors (IMT1 and IMT1B) impair the transcription of components of the OXPHOS system and thus reduce mitochondrial metabolism and ATP level. IMT1 and IMT1B have also been shown to reduce cancer cell growth and viability in vitro and to induce a strong anti-tumor response in ovarian and colon xenograft models
[36]. Inhibition of mitochondrial proteins translation was also shown to inhibit cancer cell proliferation and induce cell death
[37][38]. For instance, tigecycline, an FDA-approved broad spectrum antibiotic, dose-dependently and specifically inhibits translation by mitochondrial-but not cytosolic-ribosome, thereby leading to oxidative stress and damage, and suppression of mitochondrial respiration
[38]. Tigecycline was also shown to increase the efficiency of cisplatin in ovarian cancer cells
[38] and of tyrosine kinase inhibitor Imatinib in chronic myeloid leukemia
[39]. A phase 1 dose-escalation study of tigecycline administered intravenously to patients with acute myeloid leukemia showed a safety profile
[40].
3.2. Targeting ETC
Several ETC inhibitors have been shown to disrupt the function of respiratory complexes of the ETC and to induce high levels of ROS that trigger cancer cell death
[41]. Most of them target the complex I and are particularly efficient in tumors that rely on OXPHOS for their survival. Promising inhibitors such as metformin, ME344 and IACS10759 are currently in clinical trials. Metformin requires organic cation transporters (OCT) to enter cells and then acts as an anticancer agent through inhibition of mitochondrial NADH dehydrogenase (complex I)
[42]. In head-and-neck and breast cancer cells, anti-tumor effects of metformin rely on the expression of OCT
[43][44], which may explain the variability of metformin efficiency in clinical trials. OCT expression may thus constitute an appropriate predictive biomarker to identify tumors that are likely to benefit from metformin therapy
[45]. Compound ME-344 is a second-generation isoflavone that inhibits mitochondrial NADH biquinone oxidoreductase of complex I
[46]. ME-344 also generates ROS, leading to the translocation of Bax to the outer membrane. This translocation induces mitochondrial permeability transition which favors the release of pro-apoptotic molecules
[47]. ME-344 was shown to reduce cell growth and viability of AML cell lines and primary AML patient samples, with no effect on normal hematopoietic cells
[48]. In a randomized phase 0/1 trial, ME-344 displayed a significant anti-tumor activity on HER2-negative breast tumors
[49]. IACS010759 is a small molecule inhibitor that binds a subunit of complex I of the ETC to inhibit electron transfer. IACS010759 efficiency is under evaluation in clinical trials on AML and advanced solid tumors
[50][51]. It is to note that in tumors with intact glycolytic system, ETC inhibition may increase glycolysis as an adaptive metabolic response to counteract reduced ATP production and may account for resistance to ETC inhibitors. Thus, combination of IACS10759 with an inhibitor of glycolysis such as 2-Deoxy-glucose (2DG) may represent a useful combination to prevent resistance
[52].
3.3. Targeting the TCA Cycle
The TCA cycle provides all the biosynthetic precursors necessary for cancer cell growth and maintenance. Indeed, inhibitors of TCA cycle enzymes have shown anti-cancer potential in several cancer types. Among them, CPI-613 (Devimistat) is a lipoate analog that inhibits both α-ketoglutarate dehydrogenase (OGDH) and PDH to prevent the entry of glucose or glutamine-derived carbons into the TCA cycle and alter redox homeostasis
[53]. After a safety phase 1
[54], CPI-613 has shown promising results in combination with cytotoxic chemotherapy in a phase 2 study on relapsed or refractory small cell lung carcinoma
[55] and in a phase 3 study on metastatic pancreatic adenocarcinoma
[56]. Inhibitors of defective enzymes, such as IDH, responsible for 2-HG oncometabolite accumulation, have also been developed. AG221 (Enasidenib) and AG120 (Ivosidenib) efficiently reduce the level of 2-HG and were FDA-approved for IDH-mutated relapsed or refractory acute myeloid leukemia
[57]. Inhibition of enzymes that provide pyruvate and glutamate to the TCA cycle are currently under investigation. Targeting glycolysis, either by inhibition of glucose transporters or hexokinase 2, or by using glucose analogs that cannot be metabolized, showed promising results in preclinical studies. However those strategies gave negative results in clinical trials, due to either high toxicity or lack of efficiency
[45][57]. Glutamine pathway contributes to ATP production and protein synthesis but also to the control of ROS homeostasis. As for glucose analogs, glutamine analogs have shown severe toxicities, therefore therapeutic strategies have mainly focused on glutaminase (GLS) inhibition. Several inhibitors have been positively evaluated in preclinical studies, showing reduced tumor growth of soft tissue carcinomas, triple negative breast cancers and hematological tumors
[58][59][60] and one of them, CB-839 (Telaglenastat), has been evaluated in phase 1/2 trials. A discovery program focusing on optimizing the physicochemical and pharmacokinetic properties of GLS inhibitors has recently been launched
[61]. Compound 27 (IPN60090, derived from CB-839) was identified as an orally available and efficient compound in xenograft models and is currently in phase 1 clinical trial
[61].
3.4. Targeting Redox Homeostasis
Pharmacological increase of ROS level over a toxic threshold has been assessed as cancer therapy. Therapeutic strategies include increased production of ROS or reduced antioxidant response. Indeed, cytotoxic chemotherapies such as cisplatin, 5-Fluorouracil or paclitaxel promote a high level of oxidative stress by increasing ROS. Procarbazine was the first ROS-producing anticancer drug and is approved for the treatment of brain tumors and lymphomas
[62]. Elesclomol (STA-4783) is another ROS-generating compound in phase 2 trials for malignant mesothelioma, metastatic melanoma, prostate cancer, advanced kidney cancer and resectable esophageal cancers
[62][63]. Elesclomol inhibits super oxide dismutase SOD1 and thus increases ROS by impairing antioxidant defense. Interestingly, delivery of drugs directly into the mitochondria disrupts mitochondrial function and induces mitochondria-dependent apoptosis via rapid generation of ROS
[64]. Methods to selectively target mitochondria include the coupling of lipophilic cation such as triphenylphosphonium group (TPP+) to anticancer drugs. Increased mitochondrial transmembrane potential observed in cancer cells favors preferential accumulation of TPP conjugated drugs into cancer cell mitochondria. This new generation of compounds, named ‘mitocans’, efficiently kill multiple types of cancer cells. For instance, mitochondrial targeted vitamin E succinate targeting complex II (MitoVES) and mitochondrial targeted tamoxifen targeting complex I (MitoTAM) efficiently kill colorectal, lung and breast cancer cells and inhibit tumor growth by interfering with complex I-/complex II-dependent respiration without systemic toxicity. Promising results were obtained with MitoTAM tested in phase 1 trial, that is currently extended to phase 2. The promising mitochondrial targets in cancer therapy are presented in
Figure 2.
 +1 point
+1 point