1000/1000
Hot
Most Recent
 +1 point
+1 point
There are different types of anti-inflammatory agents, including small molecules, peptides, and antibodies. In this entry, there will be a focus on small molecules for anti-inflammatory treatments as they have been the center of traditional medicine. Small molecule drugs are compounds with low molecular weight that can easily enter the body and modulate biochemical processes to treat medical conditions.
Although the mechanism is not yet fully understood, it is believed that reactive oxygen species, free fatty acid intermediates, and adipose tissue dysregulation promote inflammation through high levels of proinflammatory adipokines and low levels of anti-inflammatory adiponectines Zebrafish present receptors, mediators, and inflammatory cells similar to mammals and humans making them suitable animal models to study new anti-inflammatory agents and their mechanisms [1]. There are different types of anti-inflammatory agents, including small molecules, peptides, and antibodies. In this review, there will be a focus on small molecules for anti-inflammatory treatments as they have been the center of traditional medicine.
Recent investigations have found novel anti-inflammatory agents from natural sources, including cyanobacterial extract [2], a norditerpenoid from the Hainan soft coralSinularia siaesensis[3], and peptides extracted from the adzuki bean [4]. Willow bark is considered one of the first examples of modern drug development from herbal plants. The effectiveness and safety profile of herbal medicines such as willow bark extract is of great interest. The action of willow bark extract as an anti-inflammatory agent was compared to celecoxib and acetylsalicylic acid, and it was found to be as effective as these drugs [5].
The main target of anti-inflammatory drugs (NSAIDs) is the cyclooxygenase (COX) pathway composed of two isoforms of the enzyme, i.e., cyclooxygenase-1 (COX-2) and cyclooxygenase-2 (COX-2). Both isoforms have an alpha carbon RMSD of only 0.9 Å. Cyclooxygenases are key in the lipid signaling pathway, being the first and rate-dependent step in prostaglandin and thromboxane synthesis [6][7]. Therefore, NSAIDs work by inhibiting the COX pathway and preventing the synthesis of prostaglandins [8]. In the body, arachidonic acid (AA) is transformed by COX-1 and COX-2 in lipid signaling to prostaglandin and thromboxane to mediate inflammatory processes [9].
Functions of prostaglandin other than mediating the inflammation processes are to protect the gastric mucosa and stimulate platelet aggregation. Therefore, reversible COX-1 inhibition may be used as an antiplatelet agent helping patients with cardiovascular conditions when aspiring is not sufficient. Selective COX-1 inhibitors may be safer than nonselective inhibitors associated with higher risks of upper gastrointestinal bleeding [10] and selective COX-2 inhibitors that are linked to cardiovascular effects [11][12][13].
The active site of COX-1 enzyme is formed by Val116, Arg120 Its difference with the COX-2 active site is the presence in COX-2 of Ile523 instead of Val509 and Arg499 instead of His513, which makes the COX-2 active site 20% bigger and with a hydrophilic side chamber [14][15][16]. COX-2-selective inhibitors take advantage of the side pocket and bind in a different mode compared to nonselective compounds, making an additional salt bridge and three extra hydrogen bonds [6]. NSAIDs inhibit COX enzymes in a reversible competitive manner or in a slow tight-binding way depending on the speed and efficiency of each molecule in displacing water molecules inside the pocket and forming hydrogen bonding [17].
Structural and functional information about COXs has been widely studied, identifying structural features key for binding [7] and using different techniques such as saturation transfer difference NMR spectroscopy (STD) [18]. While the aromatic ring makes hydrophobic interactions with the pocket, the acidic group forms hydrogen bonds with Arg120 and Tyr355 [6]. In the study of Viegas et al., known COX-1 and COX-2 inhibitors were evaluated, finding a good correlation between experimental crystallographic structure and STD signal [18]. Ibuprofen, diclofenac, and ketorolac bind in a similar way to COX-2, where the ligand moieties form tighter interactions towards Arg-120 and Tyr-355 than towards Ser-520 and Tyr-385 [18].
Some types of cancer such as epithelial ovarian cancer are reported to overexpress COX-1. In this scenario, selective COX-1 inhibitors may be useful as clinically proven to detect cancer in an early state (imaging agents) but also as therapeutic agents [19]. The link between inflammation and cancer occurs in the inflammatory pathway and includes proinflammatory agents such as cytokines. Studies have also shown upregulation of COX-2 during cancer, and COX-2 has been associated with some neurodegenerative diseases [20][21].
The most prescribed family of anti-inflammatory drugs are NSAIDs, accounting for 5–10% of total prescriptions [22][23][24][25]. Some of them are over-the-counter drugs, which increases their usage [26]. NSAIDs are the most cost-effective initial therapy for inflammation and pain relief, including sports injuries, arthritis and headaches [27][28][29]. Although NSAIDs are somewhat effective for spinal pain and other acute painful conditions, there is an urgent need for new therapies to treat these medical conditions [30][31].
NSAIDs’ most common side effects include renal, hepatic, gastrointestinal, and cardiovascular reactions [26][31][32][33][34]. This side effect is mostly attributed to inhibition of COX-1, although there are several other factors involved such as the interaction of NSAID with phospholipids [26]. To ameliorate the GI effect, gastroprotective drugs like proton pump inhibitors are used together with NSAIDs [31]. In this sense, more than improving the potency of NSAIDs, what is important regarding this type of drug is to enhance gastrointestinal safety [14][25].
Ibuprofen belongs to the family of propionic acid derivatives. Its main difference from other propionic acid derivatives lies in its pharmacokinetic characteristics [35]. It is used in antirheumatic treatments, sports injuries, and menstrual cramps. As silicon is more electropositive than carbon, hydrogen bonding can be enhanced [36].
Some of the drawbacks of ibuprofen’s properties are its low solubility in physiological media, high melting point, and high melting enthalpy, which make the production of intravenous formulations challenging [37]. The solubility of sila-ibuprofen is enhanced, going from 21 mg/L for ibuprofen to A difference in the electrostatic potential is also noticed, as it changes from positive to negative around the silicon atom [37]. Free energy perturbation and experimental results indicate COX-1 and COX-2 inhibition, and a low toxicity profile of the drug was maintained [37].
Sodano and coworkers synthesized and evaluated a paracetamol–galactose conjugate as a prodrug [38]. In this regard, the conjugate presented higher stability in human serum and reduced in vitro metabolism, as the conjugate after was able to be found after 2 h, which does not happen when using paracetamol alone. As paracetamol is slowly released from the conjugate, a longer analgesic effect was found after oral administration lasting up to 12 h being able to treat neuropathic pain such as hyperalgesia. Moreover, the hepatotoxicity was significantly reduced compared to paracetamol [38].
Changing functional groups is also an interesting approach to increase the safety profile of a drug. Some studies suggest that carboxylic acid is key for binding, but it is also linked to gastrotoxicity. As oxetane is a carbonyl isostere, carboxylic acid may be exchanged for oxetan-3-ol. Furthermore, linking furoxan and furazan groups with ibuprofen produces compounds2and3(Figure 1) with better gastrotoxicity properties without changing the anti-inflammatory effect [39].
This approach has been applied to find new treatments for Alzheimer’s disease [40], malaria [41][42], diabetes [43][44], cancer [45][46][47], and HIV [48]. Based on the anti-inflammatory activity of pyrazolo[1,5-a]pyrimidine, 2,5-diarylpyrazolo[1,5-a]pyrimidin-7-amines, new compounds were synthesized. Eleven compounds (4–14;Figure 1) showed interesting anti-inflammatory properties compared to indomethacin (Table 1) [49]. For R’, it was observed that H and F diminished activity while Cl, CH3, and Br increased it.
| Compound | Inhibition (%) |
|---|---|
| 4 | 54.76 |
| 5 | 51.16 |
| 6 | 66.66 |
| 7 | 34.52 |
| 8 | 64.28 |
| 9 | 80.95 |
| 10 | 67.85 |
| 11 | 76.19 |
| 12 | 65.47 |
| 13 | 66.66 |
| 14 | 64.28 |
| Indomethacin | 84.52 |
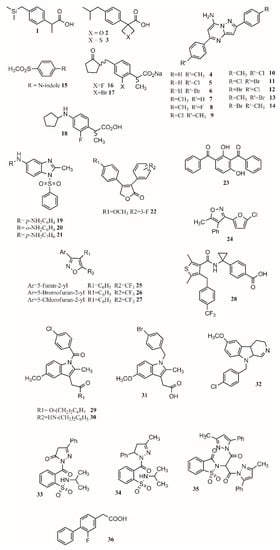
In another study, Harrak et al. designed 4-(aryloyl)phenyl methyl sulfones as anti-inflammatory compounds against COX-1 and COX-2 [27]. Molecular modeling results showed how the methylsulfone group in the studied compounds fit the COX-2 pocket, making hydrogen bonds with Arg120, Ser353 and Tyr355.N-Arylindole (15;Figure 1) was found to be the most potent and selective COX-2 inhibitor (COX-1/COX-2 ratio of 262), having greater anti-inflammatory activity than ibuprofen in vivo [27]. The pose of the ligands allows forming hydrogen bonds with Arg120, Ser353 and Tyr355 and van de Waals contacts with Val349, Therefore, the conjunction of a methylsulfone and an aryl group is enough to produce interesting anti-inflammatory activity [27].
proposed that gastric lesions are related to membrane permeabilization. Therefore, loxoprofen derivatives with lower membrane permeabilization should produce fewer gastric lesions. Synthesized compounds16and17(Figure 1) have lower membrane permeabilization and indeed produced fewer gastric lesions compared to loxoprofen but with equivalent activity [22]. Other types of compounds that possess low ulcerogenicity compared to indomethacin are 5-substituted-1-(phenylsulfonyl)-2-methylbenzimidazole derivatives, where compounds19,20and21(Figure 1) also presented good anti-inflammatory properties in in vivo assays, making them good anti-inflammatory candidates [50].
It was found that different positions of different functional groups such as trifluoromethyl, halogens, phenoxy, alkyl, alkoxy, and thioalkyl influence COX inhibition and potency. Another approach to find COX-1 inhibitors is the use of protein affinity fingerprints. Hsu and coworkers identified the fingerprint of 19 COX-1 inhibitors and developed a model to screen 62 compounds for new possible COX-1 molecules [51]. Interestingly, although a carboxylate group is present in known COX inhibitors such as ibuprofen or naproxen, that functional group was not found in newly identified COX-1 inhibitors.
Vitale et al. found that 3-(5-chlorofuran-2-yl)-5-methyl-4-phenylisoxazole (24;Figure 1) is a potent COX-1 inhibitor, where the isoxazole ring and the furanyl group are key for selectivity [10]. Furthermore, a SAR series based on this compound was performed, showing that the incorporation of 5-chlorofuran-2-yl, 4-phenyl and 5-methyl groups on the isoxazole core enhances selective inhibition of COX-1. Two extra changes were shown to increase potency with a slow reversible process: exchanging chlorine with bromine or a methyl group in the furyl core and placing a CF3group instead of the 5-methyl group (25,26,27;Figure 1). Furthermore, compound25was shown to be >1000-fold more potent for COX-1 than COX-2, having a slow reversible interaction with the first one and a fast one with the second [10].
Starting from known selective COX-2 inhibitors and after elucidating their mode of binding by X-ray crystallography or NMR, a pharmacophore model can be built in order to identify the key features for binding and design new molecules that can inhibit COX-2 selectively. The basis of selective inhibition lies in the large Ile523 on the entrance of the side pocket which prevents bulk and rigid functional groups such as the sulfamoyl or sulfonyl from interacting with the COX-1 side pocket [14]. Furthermore, synthesizing amides and esters of known inhibitors such as meclofenamic acid [52] and indomethacin [53] resulted in compounds with interesting COX-2 inhibition properties. In this sense, it has been shown that indomethacin esters and amides are slow, tight-binding COX-2-selective inhibitors eliminating gastric side effects.
Compound33(Figure 1) presents an activity of 0.09 (SI ¼ 135.9); exchanging the benzene ring with a methyl group to form34(Figure 1) increased activity and selectivity to 0.06 mM (SI ¼ 154), and the formation of35(Figure 1) increased activity and selectivity to 0.05 mM Therefore, pyrazoles present better selectivity and activity than hydrazone. The most active benzenesulfonamides are the ones bearing dipyrazole, followed by those with pyrazole, triazole and oxadiazole, with activities similar to celecoxib and presenting a low ulcer index [28].
One of the main functions of COX-2 is to oxygenate arachidonic acid, 2-arachidonoylglycerol (2-AG), and arachidonoylethanolamide (AEA). Inhibiting the second active site requires higher concentration than inhibiting only the first one, allowing substrate-selective inhibition [54]. It is reported that COX inactive (R)-profens can inhibit endocannabinoid oxygenation but not arachidonic acid oxygenation in a substrate-selective manner. μM. Aryl groups are better than other groups, and fluriprofen is more potent than ibuprofen [54].
In a novel approach, Bhardwaj et al. used the COX-2 enzyme to produce selective inhibitors in a process called in situ click chemistry. Lead compounds are produced through a [49][55]-cycloaddition inside the active site of COX-2. The SO2Me functional group, the orientation of the azide group inside the pocket, the size, and several interactions are key for the synthesis and later inhibition of this selective compound. When adding a series of precursors, COX-2 was able to choose the one that produced the best fit, producing better inhibitors than celecoxib [20].
Sometimes, monotherapy is not considered an effective treatment in complex disorders such as cancer, diabetes, infection, or inflammatory conditions [56]. Fixed-dose drug combination is an alternative, although this type of therapy may increase the risk of adverse drug–drug interactions and produce changes in the pharmacokinetic profile of one of the components [57]. Cocrystals have emerged as compounds with improved properties and performance including solubility, incompatibility, and stability. One example is the tramadol-celecoxib cocrystal, which successfully passed phase II clinical trial for the treatment of acute pain [56][58].
[13][59], soluble epoxide hydrolase [60], phosphoinositide-3-kinase delta [61], or fatty acid amide hydrolase (FAAH) Advantages of this dual inhibition include greater anti-inflammatory effect with reduced side effects [11]. The leukotrienes produced by 5-LOX are important inflammatory mediators and may be related to cancer, cardiovascular diseases, and gastrointestinal reactions. Still, 5-LOX inhibitors do not show anti-inflammatory effects [13].
Hybrid multiligand molecules containing NSAID structures can lead to enhance anti-inflammatory activity by inhibiting different targets inside the inflammation pathway [62][63]. COX-2/5-LOX inhibitors are compounds that offer more efficacy, fewer side effects, and broader anti-inflammatory properties. Similarly, by joining β-boswellic acid with different NSAID molecules via a Steglich esterification, compounds with interesting anti-inflammatory and antiarthritic properties can be obtained [55]. All hybrid molecules have synergistic effects in inflammation and arthritis, with37and38being the best (Figure 2).
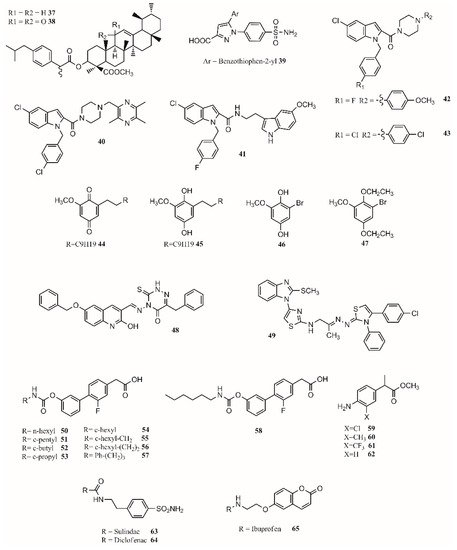
Gedawy et al. synthesized a series of pyrazole sulfonamide as anti-inflammatory agents where benzothiophen-2-yl pyrazole carboxylic acid derivative39(Figure 2) was the best lead found, with an IC50of 5.40 nM for COX-1, 0.01 nM for COX-2, and 1.78 nM for 5-LOX. Its selectivity index towards COX-2 was 344.56 [14].
The phenoxy acetamide, indole, chalcone thiosemicarbazone, and quinoline are important groups in anti-inflammatory agents [64][65][66]. Huang, Qian, and coworkers designed indole-2-amides as anti-inflammatory drugs [13][67]. Results showed that40,41,42and43(Figure 2) present interesting anti-inflammatory properties, with40being the most promising compound with COX-2 selectivity and dual COX-2/5-LOX activity binding in the same way as coxibs into the COX-2 active site [13].
Dual inhibitors can also be obtained from natural sources. Primin, a quinone extracted from Primula obconica, presents good anti-inflammatory activity. Therefore, related compounds including hydroquinone, benzoquinone, and resorcinol groups were designed and tested for activity against COX-1, COX-2 and 5-LOX. (Figure 2) were found to be dual COX/5-LOX inhibitors.
In the design of multitarget compounds,50((±)-2-[3-fluoro-4-[3-(hexylcarbamoyloxy)phenyl]phenyl]propanoic acid, ARN2508;Figure 2) was found to be a potent FAAH and COX inhibitor without the gastrotoxic effect. and53is similar to54. It seems that theN-terminal region of the carbamate group may increase the interaction with FAAH. The compound57(Figure 2) is the most active compound, showing that small and branched alkyl groups substantially affect inhibitory activity [11].
Other types of dual inhibitors are the ones inhibiting COX and carbonic anhydrase (CAI) [68]. CAI inhibitors are more effective in treating rheumatoid arthritis than common NSAIDs as they do not increase the risk of oxidative stress in patients [69][70]. Its pharmacological treatment includes the use of common NSAIDs; glucocorticoids (prednisolone); and antirheumatic agents, including aminosalicylates (sulfasalazine), antimalarial drugs (hydroxychloroquine), immunosuppressants/cytostatic drugs (methotrexate), and antirheumatic drugs (aurothiomalate) [71]. Hybrid compounds containing NSAID drugs and sulfonamides and carboxylates moieties can be synthesized as dual COX
Finally, by a covalent link between a sulohydroxamic acid moiety and known NSAIDs via a two-carbon ethyl spacer, prodrugs with anti-inflammatory activity and the ability to release nitric oxide and nitroxyl were synthesized [12]. The esters produced with naproxen and ibuprofen showed greater anti-inflammatory activity than their parent compounds, while indomethacin ester was shown to be a selective COX-2 inhibitor with no ulcerogenic effect [12]. The multitarget drugs described in this section are shown inFigure 2.
Opioids and NSAIDs are the first options to treat acute pain [72]. Still, those drugs have shown to be not as effective in treating chronic pain, which may be disabling in some cases [73]. The endocannabinoid system involved in pain and inflammation processes has emerged as a promising approach to treat chronic pain [73][74][75]. Cannabinoids acting as positive allosteric CB1 receptor modulators, CB1 agonists, CB2 agonists, and mixed CB1/CB2 agonists have been reported to help in inflammatory and neuropathic pain [76][77][78].
The cannabis plant has been cultivated for hundreds of years and used for medicinal, spiritual, and recreational purposes. The main components in the plant are alkaloids which have been demonstrated to present anti-inflammatory activity [79]. Some countries have approved the use of cannabis to treat pain, multiple sclerosis, epilepsy, sleep disturbance, and neurodegenerative diseases. Furthermore, antiseizure effects in Lennox–Gastaut syndrome and Dravet syndrome have been observed in randomized controlled trials [74][80].
The term medical cannabis has arisen in the last years and refers to the use of the cannabis plant to treat different medical conditions as prescribed by a doctor [81][82]. The main use of medical cannabis is for pain management [83]. The most accepted mechanism for the analgesic effect of cannabis is through the reduction of neural inflammation and descending inhibitory pain and the modulation of postsynaptic neuron excitability pathways. Although more than 10,000 scientific articles present “conclusive or substantial evidence” and support the use of cannabis for neuropathic pain, there is a need to carry out long clinical trials to establish its safety, dosage, and indications for medical conditions [73].
Studies have shown cannabinoids act on different targets in the peripheral and central nervous system [84][85]. Their targets include CB1/CB2 receptors, GPR55, GPR18,N-arachidonoyl glycine (NAGly) receptor, nuclear receptors, ion channels, and other potential targets in the CNS [84][86]. Furthermore, cannabinoids may also act on γ-aminobutyric acid, serotonergic, adrenergic and opioid receptors that are part of the analgesic pathway [75].
Although cannabinoids are considered the main active ingredients in cannabis, other compounds inside the plant, including terpenoids and flavonoids, may also be involved in the anti-inflammatory and analgesic effect of cannabis [87][88]. THC was first isolated in the 1960s, and it is responsible not only for the psychoactive effect but also for some of the analgesic and anti-inflammatory properties. Cannabis prohibition started in the 20th century due to its psychoactive properties (ElSohly et al., 2017). Psychotropic effects include euphoria and paranoia that occur due to the activation of CB1 receptors [73].
THC is a partial CB1 and CB2 receptor agonist, while CBD, a nonpsychoactive cannabinoid, has little affinity to both receptors. Studies have shown it is able to antagonize those receptors in presence of THC acting synergically [78]. Obtaining insight into the action mechanism, CBD is a noncompetitive negative allosteric modulator of the CB1 receptor [89]. CBD modulates non-cannabinoid G protein-coupled receptors (GPCRs), ion channels, and peroxisome proliferator-activated receptor (PPARs), affecting the perception of pain [85][90].
Inhaled cannabinoids are rapidly absorbed in the bloodstream. Gastrointestinal absorption is irregular, causing low bioavailability and poor pharmacokinetic profile, although it can be increased with food [91]. This is a limitation for using cannabis in oral formulations [73]. Nabiximols, a mixture of CBD and THC approved in Spain, Switzerland, Australia, Canada, Brazil and other countries, is prescribed to treat spasticity in multiple sclerosis [80].
In this regard, the medicinal chemistry approach towards the use of cannabinoids to treat medical conditions has focused on the modulation of the cannabinoid receptors CB1/CB2 and two endocannabinoid deactivating enzymes, monoacylglycerol lipase (MGL) and enzymes fatty acid amide hydrolase (FAAH), including allosteric inhibition [92]. Aminoalkylindole is the first different chemotype to be introduced in cannabinoid medicinal chemistry. present potent antinociceptive properties, but due to their structure, some of them have been classified as controlled substances. (68;Figure 3) is a potent CB1/CB2 receptor agonist with clinical efficacy against severe traumatic brain injury [92].
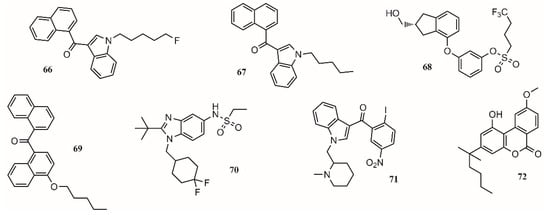
One approach to eliminate undesirable side effects is to design peripherally restricted agonists. In this sense, SAB378 (69;Figure 3) and AZD1940 (70;Figure 3) are good candidates due to their interesting properties. The discovery of selective CB2 receptors was important because their presence is peripheral, exhibiting antinociceptive and anti-inflammatory action without the CB1 side effects. AM1241 (71;Figure 3) and AM1710 (72;Figure 3) were tested in rodent models and found to be successful for treating inflammatory and neuropathic pain [92].
In 29 trials performed between 2003 and 2014, more than 75% of the studies showed a significant analgesic effect in chronic noncancer pain, including neuropathic pain, rheumatoid arthritis, and fibromyalgia [93]. The compounds targeting CB1 and/or CB2 receptors are shown in Figure 3.





