1000/1000
Hot
Most Recent
 +1 point
+1 point
Hutchinson–Gilford progeria syndrome (HGPS), or progeria, is an extremely rare disorder that belongs to the class of laminopathies, diseases characterized by alterations in the genes that encode for the lamin proteins or for their associated interacting proteins. In particular, progeria is caused by a point mutation in the gene that codifies for the lamin A gene. This mutation ultimately leads to the biosynthesis of a mutated version of lamin A called progerin, which accumulates abnormally in the nuclear lamina. This accumulation elicits several alterations at the nuclear, cellular, and tissue levels that are phenotypically reflected in a systemic disorder with important alterations, mainly in the cardiovascular system, bones, skin, and overall growth, which results in premature death at an average age of 14.5 years. Unlike the majority of the rare diseases, it has, since November 2020, a specific FDA approved drug, lonafarnib. However, this small molecule represents a treatment, but it does not cure the disease, and it has several limitations that make the development of new therapeutic strategies a critical need in the field.
Hutchison–Gilford progeria syndrome (HGPS), or progeria, is a rare human genetic disorder caused by a specific mutation in the LMNA gene, which codes for lamins A and C. This autosomal dominant disease is triggered by the single nucleotide substitution mutation c.1824C > T in exon 11. Although this mutation should not entail any consequence since both nucleotide triplets (wild type and mutant) code for glycine (p.G608G mutation), the mutation activates a cryptic donor splicing site for lamin A-specific transcript processing, leaving the splicing for the lamin C transcript unaffected (see Figure 1 for details) [1][2][3]. The appearance of this new splicing site in the lamin A gene leads to the generation of a messenger RNA with a missing part, resulting in the production of a mutated form of lamin A, which is called progerin. Progerin lacks 50 amino acid residues encoded by the lost fragment in exon 11. This deletion is critical because it contains a recognition site for ZMPSTE24 cysteine proteinase, which is involved in the last step of the post-translational processing of prelamin A protein. In normal cells, prelamin A, as expected from the CAAX sequence in its C-terminus, is subjected to a specific set of post-translational modifications. These transformations include the following stages: (i) farnesylation of the cysteine residue; (ii) cleavage of the AAX terminal tripeptide; (iii) carboxymethylation of the C-terminal cysteine; and (iv) final excision of the last 15 amino acids due to the existence of a specific recognition site. The first step is catalyzed by the enzyme farnesyl transferase (FTase), the two hydrolytic events are mediated by ZMPSTE24, and the methylation reaction is carried out by isoprenylcysteine carboxylmethyltransferase (ICMT). In progerin, once the methylation step has occurred, the last peptide removal cannot occur due to the lack of the ZMPSTE24 recognition site, so the produced protein is a 15 amino acid longer, permanently farnesylated and methylated version of lamin A (Figure 1) [4].
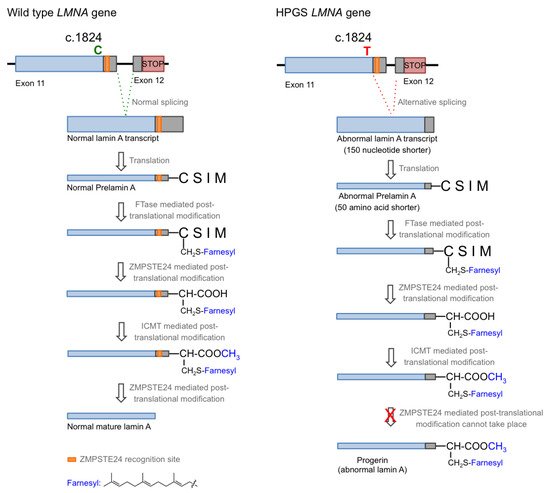
Figure 1. Biosynthesis of normal mature lamin A (left) and progerin (right).
HGPS belongs to a broader group of diseases called laminopathies [5], which are a consequence of different mutations in the LMNA gene. All of them result in a wide spectrum of overlapping disorders that comprise muscular dystrophies, a peripheral neuropathy, lipodystrophy syndromes, and accelerated aging disorders that share some of the features of progeria [6]. In addition, there are other progeria-related phenotypes referred to as non-classical mutations that are frequently described as progeroid laminopathies or atypical progeroid syndromes. These disorders have been described in detail elsewhere [7][8][9], so in this review, we will focus on classical HGPS (p.G608G mutation), which has only an autosomal dominant mode of inheritance and a clearly defined molecular background.
The increase in hydrophobicity triggers the abnormal accumulation of progerin in the nuclear membrane. This leads to the appearance of many cell defects, especially nuclear alterations, as would be expected from the fundamental structural role that lamin A plays in the nucleus. Progeroid cells display anomalous nuclear morphology and impaired nuclear functions, fundamentally due to the lack of the dynamic movement that characterizes the healthy lamin, which shifts between the nuclear lamina polymer in the nuclear lamina and the nucleoplasm [10]. Progerin, however, is strongly attached to the lamin because of its high hydrophobicity, and it is not able of participate in this dynamic cycle. This fact provokes a thickening of the lamina, enlarges cellular stiffness, and impairs many nuclear functions [11][12]. Increased cellular rigidity seems to be central in HGPS, as the disease affects mainly cells that must respond to changes in mechanical stress such as vascular cells, bone, and joints, and these three tissues recapitulate some of the most prominent disease symptoms in progeria patients [13][14]. Other remarkable nuclear defects in HGPS cells are related to DNA and chromosome functions. They include extensive alterations in chromatin structure, as reflected in the loss of heterochromatin domains, changes in epigenetic markers, and reduced DNA repair mechanisms [15][16]. These deficiencies translate into global cell defects that include chronic p53 signaling, altered inflammatory response, metabolic changes, autophagy deregulation, and stem cell dysfunction [2][17]. Altogether, they are eventually responsible for the complex and distinctive disease phenotype, characterized by growth impairment, low body weight, absence of subcutaneous fat, lipodystrophy, decreased joint mobility, alopecia, and cardiovascular disease, features that reflect in the overall premature aged aspect of diseased children (see Figure 2 for an schematic summary). Average life expectancy is about 14.6 years, and the main direct cause of death is cardiovascular disease (CVD) characterized by atherosclerosis, vascular stiffening and calcification, electrocardiographic alterations, and left ventricular diastolic dysfunction, and derived complications [14][18].
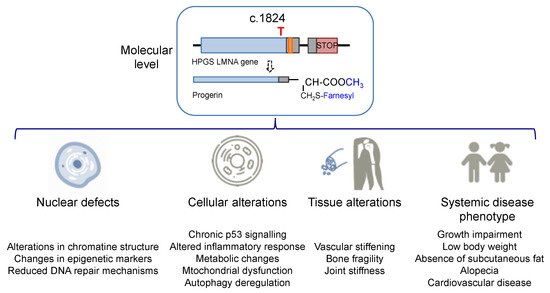
Figure 2. Alterations in HGPS individuals from the molecular and cellular levels to the disease phenotype.
In the normal post-translational prelamin A maturation process, the cytosolic enzyme FTase adds a 15-carbon farnesyl lipid to prelamin A that is eventually removed after the four-step translational processing suffered by this protein, schematized in Figure 1. However, the presence of the mutation prevents the last hydrolytic cleavage (Figure 1) yielding permanently farnesylated progerin. The presence of this group increases the hydrophobicity of the protein, thus promoting strong interactions with the nuclear membrane, where its abnormal accumulation severely affects the normal nuclear functions. Early studies described that blocking or preventing farnesylation improved nuclear abnormalities in mouse and human progeroid fibroblasts and reduced nuclear blebbing and the number of misshapen nuclei [11][19][20]. Furthermore, in vivo treatment with an FTase inhibitor (FTI) ameliorated the progeria phenotype in the Zmpste24-deficient mouse model of progeria [21]. Accordingly, inhibition of FTase was one of the first therapeutic strategies suggested for ameliorating the severity of the disease [22] and the only one that, up to date, has allowed the first approved drug for the specific treatment of progeria to be developed. FTIs were initially developed as anticancer compounds, aimed at preventing the permanent Ras activation characteristic of the frequent and deadly Ras-driven tumors [23]. The rational of this therapeutic potential was based on the fact that in the absence of farnesylation, Ras would be unable to attach to the cell membrane [24]. Hence, in the presence of an FTI, Ras should be inactive. Although the lack of efficacy of FTIs in phase III clinical trials stopped their advance into the clinic, the similarity between the post-translational processing of progerin and Ras (both belong to the CAAX family of proteins) set up the bases for the repurposing of FTIs for treating progeria by reducing the levels of farnesylated progerin. Among the different FTIs showing high efficacy in the inhibition of FTase in vivo, lonafarnib (Figure 3) soon stood out as the most promising candidate to start a single-arm phase II clinical trial in 2007 (NCT00425607) [25]. This trial showed that lonafarnib was well tolerated, and it also showed the capacity of this compound to improve some of the symptoms of the disease, such as rate of weight gain, arterial pulse wave velocity, carotid artery echodensity, skeletal rigidity, and sensorineural hearing. Importantly, it also decreased mortality rate (3.7% vs. 33.3% after a median of 2.2 years of follow-up in individuals receiving lonafarnib monotherapy compared with no treatment) [26]. Although lonafarnib does not correct all the alterations of the disease, such as lipodystrophy, skin features, alopecia, and joint contractures, the fact that it is the only available drug for treating this lethal pathology and that it has a positive impact in ameliorating specific features of HGPS, has led to its recent approval by the FDA under the name of ZokinvyTM [27].
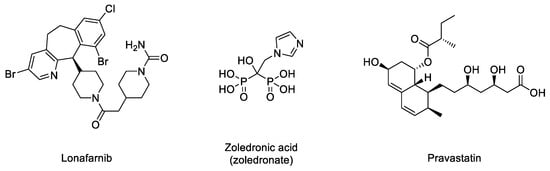
Figure 3. Structures of lonafarnib, zoledronic acid, and pravastatin.
There may be various reasons for lonafarnib’s limited improvement. One of the first hypotheses suggested that in the absence of FTase activity, alternative prenylation by the enzyme geranyl geranyltransferase (GGTase) could take place, as this mechanism had been already characterized in the limited Ras inactivation observed in vivo after treatment with FTIs. Hence, it was suggested that the concomitant inhibition of both enzymes could increase the efficacy of the treatment. The phenotypic improvement observed in an in vivo model of progeria after the combination of the statin pravastatin and the aminobisphosphonate zoledronate seemed to confirm this idea [28] and was the basis for starting a triple therapy clinical trial in which lonafarnib, pravastatin, and zoledronic acid were administered to progeria patients (clinical trials NCT00879034 and NCT00916747). Although no participants withdrew because of side effects, no significant improvements other than increased bone mineral density were observed compared to lonafarnib monotherapy. Hence, as the triple therapy did not convey any obvious advantage over monotherapy, it was discontinued, and the clinical trial was prolonged but only with lonafarnib administration [29]. In addition, it is possible that other targets of FTase, apart from progerin, are affected by lonafarnib, eliciting adverse effects in progeroid patients. In line with this concern, since lonafarnib was originally developed for the treatment of Ras-dependent tumors [24], it is antiproliferative, a feature that can potentially limit its positive effects on progeroid cells, in which pro-proliferative effects are needed. In this regard, it has been reported that after long-term treatments, lonafarnib induced cell death and formation of donut-shaped nuclei [30][31]. These negative effects should be taken into consideration, especially when analyzing the effects of combining lonafarnib with other potential anti-progeroid therapies [31].
Considering the importance of the farnesyl and methyl groups in increasing the lipophilicity of progerin and the efficacy of the FTI lonafarnib to improve the phenotype of the disease, the possibility that reducing methylation could reduce the abnormal accumulation of progerin in the nuclear membrane and hence improve the phenotype of the disease has received increasing attention. The importance of carboxyl methylation for proper membrane localization has been already described for other CAAX proteins, such as Ras [32][33]. In addition, inhibition of ICMT has been described as a promising strategy to inactivate Ras by inducing its mislocalization from the cellular membrane [34][35][36]. In this context, the finding that genetic disruption of ICMT improved the phenotype in the ZMPSTE24 mouse model of progeria supported that a small molecule ICMT inhibitor could represent a new therapeutic strategy to address HGPS. Within this aim, recent studies have provided strong evidence for the potential of ICMT inhibitors to improve the disease phenotype both in cellular and animal models. In particular, an optimized ICMT inhibitor, UCM-13207 (Figure 4), was able to increase cell viability, delocalize progerin from the nuclear membrane, and decrease DNA damage in cells from progeroid mice as well as in human fibroblasts from HGPS patients. In addition, this compound showed excellent efficacy in the in vivo progeroid mouse model LmnaG609G/G609G, where the compound increased body weight, enhanced grip strength, extended lifespan by 20%, and decreased tissue senescence in different organs together with key cardiovascular hallmarks [37][38]. In further support of the potential of this strategy, another ICMT inhibitor, compound C75 (Figure 4), was able to delay senescence and stimulate proliferation of human HGPS fibroblasts and to mislocalize progerin from the nuclear membrane towards the nucleoplasm, although no in vivo efficacy data were reported, probably due to limited pharmacokinetic properties of the compound [39]. Together, these results support the potential of the ICMT inhibitors, by themselves or in combination therapies, for treating progeria.
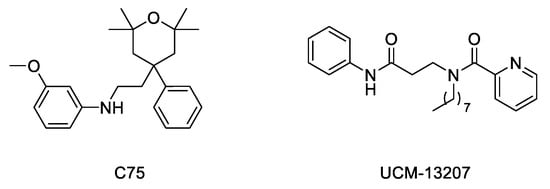
Figure 4. Structure of ICMT inhibitors C75 and UCM-13207.
Interaction between progerin and lamin A has been established as critical for the development of the senescence phenotype associated with HGPS cells. Hence, it is conceivable that an inhibitor of this protein–protein interaction could improve the progeroid phenotype. Within this aim, an ELISA-based screening was carried out to identify progerin–lamin A interaction inhibitors. This first study succeeded in the characterization of JH4 (Figure 5) as a potent and specific inhibitor of this interaction. This compound improved nuclear deformation and senescence markers of progeroid cells. Remarkably, its administration (10 mg/Kg, intraperitoneally, twice a week) to LmnaG609G/G609G progeroid mice significantly ameliorated some of the characteristic features of progeria, including increase of body weight, grab strength, skin thickness, and improved nuclear deformation. In addition, administration of JH4 was able to extend lifespan in progeroid mice and to restore senescence-related markers [40]. However, the compound half-life after oral administration was very short. Therefore, the search for new inhibitors of the interaction between progerin and lamin A with optimized pharmacokinetic properties continued until the identification of SLC-D011 or progerinin (Figure 5), a compound able to extend the life span of LmnaG609G/G609G progeroid mice after oral administration (50 mg/Kg, daily) and to improve histological and physiological hallmarks of progeria in Lmna+/G609G mice [31][41].
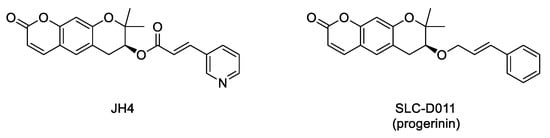
Figure 5. Structure of the inhibitors of the interaction between progerin and lamin A.
Beyond targeted therapies, alternative approaches have addressed the search of small molecules able to ameliorate or reverse the phenotypic harmful effects elicited by the abnormal progerin accumulation in the nuclear lamina. In this regard, and aimed at improving the characteristic nuclear architecture defects of progeroid cells, the development of inhibitors of N-acetyltransferase 10 (NAT10) has been explored [42]. In particular, oral administration (100 mg/kg, daily dose) of remodelin (Figure 6), a NAT10 inhibitor that acts in a progerin and FTase independent manner, significantly enhanced the health span of progeroid mice and slowed down the body-weight loss. In addition, remodelin treatment corrected several progeroid hallmarks such as the levels of subcutaneous adipose tissue and the adventitial fibrosis of the aorta. It also reduced the vascular smooth muscle cell loss both in the aorta and the coronary arteries and decreased some of the markers of genome instability in heart and lung of treated progeroid LmnaG609G/G609G mice [43].
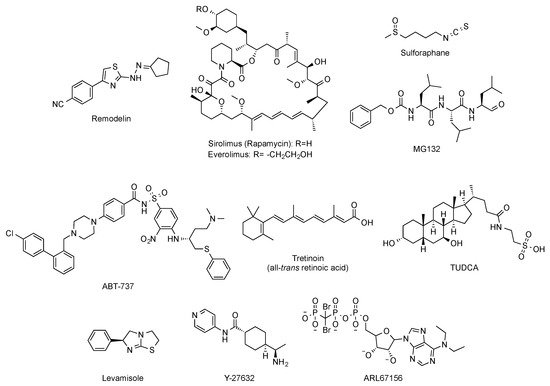
Figure 6. Structure of remodelin, sirolimus (rapamycin), everolimus, sulphoraphane, MG-132, ABT-737, tretinoin (all-trans retinoic acid), TUDCA, levamisole, Y-27632, and ARL67156.
Other strategies have considered the possibility of enhancing the autophagy-mediated progerin clearance. In this way, the use of rapamycin (Figure 6) improved the abnormal nuclear shape, delayed the onset of cellular senescence, and rescued the chromatin phenotype of HGPS fibroblasts [44][45]. This finding led to the consideration of the inhibition of the mTOR (mammalian target of rapamycin) pathway as a pharmacological strategy worth exploring for treating progeria. Aimed at this objective, a variety of studies have established that rapamycin administration (8 mg/kg, intraperitoneal) inhibits mTOR and increases autophagy. The regulation of these signaling pathways translates in vivo into an improvement in cardiac function and in lifespan extension in lamin A/C-deficient mice [46]. These results have been the basis of the phase I/II clinical trial (NCT02579044) of everolimus (Figure 6), a structurally-related analogue of rapamycin, in combination with lonafarnib, which is still ongoing. Other attempts to increase autophagy to promote progerin degradation have explored a variety of molecules such as sulforaphane or MG-132 (Figure 6) [47][48][49].
Global correction of progeroid cellular defects has been also attempted by using molecules with more general mechanisms of action such as antioxidants, reactive oxygen species (ROS) scavengers, and anti-inflammatory or senolytic compounds. For example, the capacity of the ROS scavenger N-acetyl cysteine to improve nuclear damage and cell proliferation of HGPS fibroblasts has been reported [50], and analogous effects were observed after administration of the associated protein kinase (ROCK) inhibitor Y-27632 (Figure 6) [51]. Similarly, the use of antioxidants able to improve mitochondrial function has also positive effects in progeroid cells [52]. Additionally, the selective removal of senescent cells with the senolytic compound ABT-737 (Figure 6) has a positive impact on the global senescent signature and in the median survival, which is significantly increased in the heterozygous Lmna+/G609G progeroid mouse model treated with this compound during their second half of life [53].
Phenotypic drug discovery has been also applied to progeria. In this regard, a high-content imaging-based high-throughput screening of hundreds of FDA approved molecules has been carried out, aimed at identifying those compounds that produce an improvement in the structural, epigenetic, DNA damage, and related nuclear defects characteristic of progeroid cells. This study has characterized some retinoids as another class of drugs that can be useful for treating some of the symptoms of the disease [54]. The mechanism of action of these compounds could be mediated by their interaction with the retinoic acid receptor element (RARE) present in the LMNA promoter, thereby repressing lamin A, progerin, and lamin C expression at the mRNA level. In support of this idea, the capacity of all-trans retinoic acid (Figure 6) to decrease the levels of progerin has been reported [55]. Although all these results lay the foundations for deeper studies, in general, up to this moment, the positive effects have been only assessed in cellular models, and the specific molecular targets involved are difficult to determine [56]. The objective of identifying the pathways linked to specific progerin-induced alterations has been successfully addressed in the case of VSMC damage. A recent transcriptomic study has shown that endoplasmic reticulum (ER) stress and the unfolded protein responses play a critical role in the VSMC death characteristic of progeria. Accordingly, administration of tauroursodeoxycholic acid (TUDCA, Figure 6), a chemical chaperon, to two mouse models of HGPS (one with ubiquitous progerin expression and other with VSMC-specific progerin expression) was able to decrease medial VSMC loss and atherosclerosis. In addition, in the VSMC-specific model, TUDCA also increased lifespan [57]. Additional studies have been also focused on the specific correction of some of the most deleterious symptoms of progeria, with a special attention to cardiovascular disease. In this context, reducing vascular calcification by increasing the levels of ATP and pyrophosphate with the administration of the tissue nonspecific alkaline phosphatase (TNAP) inhibitor levamisole and the ectonucleoside triphosphate diphosphohydrolase (eNTPD) inhibitor ARL67156 moderately extended (12%) longevity in the progerin-expressing LmnaG609G/G609G mouse model [58]. In the same line, dietary magnesium supplementation reduced calcification of vascular smooth muscle cells in vitro and in vivo and improved the longevity of LmnaG609G/+ mice [59]. However, the relevance of this finding to human HGPS remains to be addressed.





