1. Introduction
The COVID-19 pandemic has caused nearly 1.28 million deaths worldwide (as of 6 December 2020)
[1]. The disease has affected many human sectors worldwide and prompted scientists to explore the topic more extensively. Consequently, the number of scientific publications related to COVID-19 has increased sharply since 2020. Several bibliometric studies focused on the COVID-19 literature and aimed to understand the knowledge flow and trends
[2]. However, there has not been much research exploring in-depth knowledge unit analysis (e.g., biological entities-level explorations), especially relationships between knowledge units. This limitation might prevent us from detecting the full knowledge flow in studies. Therefore, we require solid knowledge representation with clear definitions of knowledge unit relationships for greater understanding.
This study proposes a framework to merge two independent datasets Analytical Graph (AG) and CORD-19
[3] (with significant knowledge overlaps) into a new, larger knowledge graph (KG). We build the KG to promote more profound knowledge retrieval and in-depth knowledge mining. AG is a subgraph generated from multiple biomedical KGs, while CORD-19 contains coronavirus-related scientific publications. We adopt PubTator
[4], a popular biological entity extraction tool, to extract entities from the CORD-19 literature. However, we cannot explicitly capture the relationships among extracted knowledge units. Therefore, we use AG to enrich the relationships among entities, yielding a more comprehensive, global-wise knowledge base, covering coronavirus entities and their “contexts”.
The new KG contains at least 21,700 genes, 2500 diseases, 94,000 phenotypes, and other biological entities (e.g., compound, species, and cell lines). We use 27 types of relationships in our KG and natural language processing techniques, such as entity recognition, semantic disambiguation, and knowledge merge. This KG could be used widely in the future. It functions as an essential knowledge base for related scientific research and development, while benefiting from knowledge retrieval and in-depth knowledge mining. This new KG acquires and integrates coronavirus-related information into an ontology and enables researchers to apply reasoning to derive new knowledge according to defined rules.
We evaluated the KG’s usability for information extraction using our pathfinding framework, which retrieves several paths with different depths. It calculates the path score based on the similarity distance between two nodes in every relationship found in the path. First, we transform nodes into vector values using a word vector transformation model. Then, we calculate the similarity distance between nodes using cosine similarity. We use a pre-trained word2vec model built using PubMed
® and PubMed Central
® (PMC) texts
[5].
2. Cases
2.1. Ego-Centered Subgraph
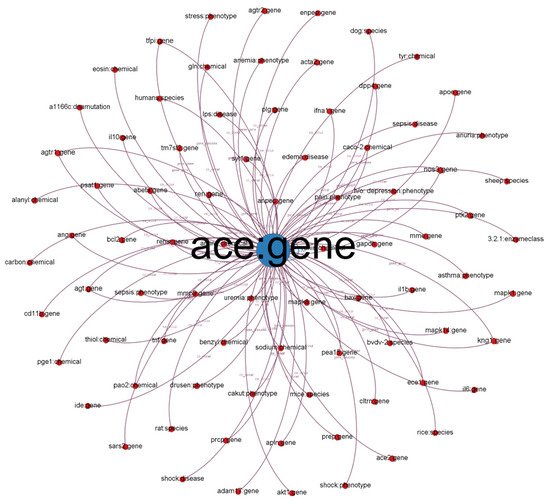
We illustrate a subgraph from our established KG in Figure 1. Each node in the subgraph represents a biological entity. Node labels represent entity names and biological types (e.g., chemical, gene, and disease). We defined different colors for different biological (node) types. The size of the nodes is proportional to their degree (the number of connected nodes). Each edge represents a relationship between two entities (e.g., co-occurrence, gene and cellular components, and gene-to-gene relationship). The label indicates the relationship type.
Figure 1. Subgraph from established KG with ACE: gene as the center node.
We chose the angiotensin-converting enzyme (ACE) gene, encoded as ACE, which has 40% overall identity to ACE-2 and is positively related to COVID-19
[6]. ACE-2 counters the related ACE activity by reducing angiotensin-II and increasing angiotensin-(1–7), making it a promising drug target for treating cardiovascular diseases. ACE-2 activators are also potential COVID-19 treatments, according to their popularity in the COVID-19 literature
[7]. As depicted in
Figure 1, we discovered that other entities such as SARS2 and PaO2 are highly related to COVID-19 because PaO2 reflects arterial oxygen tension and COVID-19 damages the lung.
2.2. Path
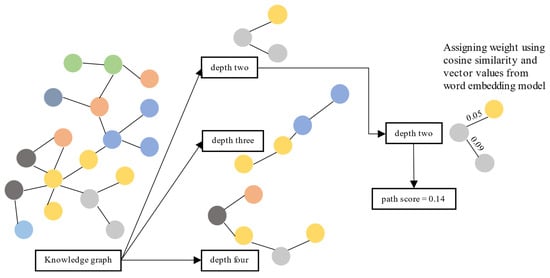
This section evaluates each path by scoring it using the similarity distance between nodes and verifying the information given in the top-ranked paths. We calculated the similarity distance using cosine similarity on vector representation from the vector transformation model
[5]. First, we retrieved the shortest paths from the source node to the target node with several depths from the KG. We used three depths: two, three, and four. Paths with a depth of two have three nodes: one source node, one target node, and one node in between. Paths with a depth of three have two nodes in between, and paths with a depth of four have three nodes.
Second, for each path, we calculated and summed the cosine similarity between nodes. Third, we measured the cosine similarity between nodes using their vector values obtained from
[5]. Finally, we sorted paths (separately for each depth) based on the sum of cosine similarity values between nodes. Then, we analyze whether the information given in the top-ranked paths is accurate. The top-ranked paths are paths with the top 95 percentile score from the distribution. We illustrate the evaluation process in
Figure 2.
Figure 2. Evaluation process for KG usability using a path ranking framework.
In this example, we analyzed two paths: (1) between IL-6 receptor and hydroxychloroquine and (2) between STAT1 and chloroquine. IL-6 receptor and STAT1 are both related to immune systems and COVID-19. We found one path with a depth two, 202 paths with a depth of three, and 600 paths with a depth of four for IL-6 receptor and hydroxychloroquine. We also found 62 paths with a depth of two, 642 paths with a depth of three, and 435 paths with a depth of four between STAT1 and chloroquine. We found evidence of gene–gene, gene–disease, and compound–disease relationships from those paths. We analyzed the evidence found in top-ranked paths for each depth and compared them with DisGeNet
[8] and DrugBank
[9].
2.2.1. IL-6 Receptor and Hydroxychloroquine
This section discusses the characteristics of paths between two entities: IL-6 receptor and hydroxychloroquine. IL-6 is Interleukin 6, an interleukin that functions as both a pro-inflammatory cytokine and an anti-inflammatory myokine. IL-6 inhibitors may ameliorate severe lung tissue damage caused by cytokine release in patients with severe COVID-19 infections. Hydroxychloroquine is a medication used to prevent and treat malaria in areas where malaria remains sensitive to chloroquine. Other usage includes the treatment of rheumatoid arthritis (RA), lupus, and porphyria cutanea tarda (PCT).
Common side effects of hydroxychloroquine consumption include vomiting, headache, changes in vision, and muscle weakness. Severe side effects may include allergic reactions, vision problems, and heart problems. Although we cannot exclude all risks, it remains a treatment for rheumatic disease during pregnancy. Companies sell hydroxychloroquine under the brand name Plaquenil (among others).
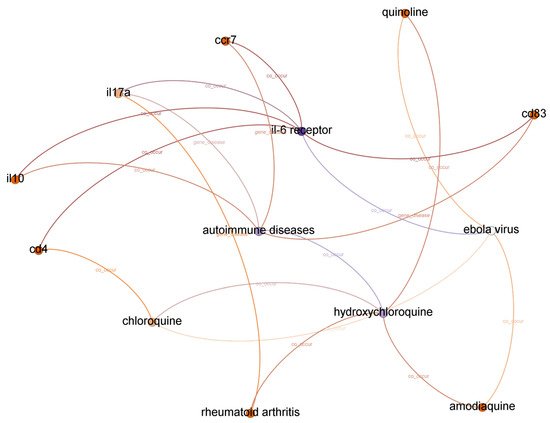
The list of top-ranked paths (top 95 percentile) with a depth of two, three, and four based on the shortest path algorithm of the graph to find and calculate the score of all paths from IL-6 receptor to hydroxychloroquine. A subgraph from top-ranked paths with a depth of three is illustrated in Figure 3.
Figure 3. Subgraph from top-ranked paths with a depth of three in IL-6 receptor and hydroxychloroquine case.
We concluded that the Ebola virus infection co-occurs with IL-6 receptor and hydroxychloroquine from the evidence found in paths with a depth of two. We assume the first relationship between IL-6 receptor and the Ebola virus is accurate because experiments in
[10] concluded that the elevated concentration of IL-6 in plasma during the symptomatic phase is a non-fatal Ebola virus infection marker. Furthermore, we found a supported argument in
[11] for the second relationship between hydroxychloroquine and the Ebola virus.
Based on the Drugbank dataset
[9], we also found that combinations with hydroxychloroquine can decrease the Ebola Zaire vaccine’s therapeutic efficacy (live, attenuated). Even though each relationship is correct, we cannot identify a potential relationship between IL-6 receptor and hydroxychloroquine from paths with a depth of two. Moreover, there is no significant relationship between the Ebola virus and COVID-19, except that both are pandemic diseases.
There are ten paths in the top-ranked path category with a depth of three. We found twenty-one different relationships (node–relationship–node). In addition to the Ebola virus, we found another disease that appeared in top-ranked paths: autoimmune diseases. We found that the relatedness between COVID-19 and autoimmune disease is more substantial compared to the Ebola virus. A recent report found autoimmune diseases in COVID-19 patients
[12]. We also found the RA disease in top-ranked paths. RA disease is related to autoimmune disease
[8], and because RA patients are more likely to catch certain infections, they have a higher chance of getting COVID-19. DisGeNet
[8] also reported that IL-6 receptor is a biomarker in RA.
We found three more compounds in top-ranked paths with a depth of three: chloroquine, amodiaquine, and quinoline. Drugbank
[9] reported that amodiaquine and chloroquine are currently in clinical trials for COVID-19. We also found several genes related to autoimmune disease or RA, such as cd4, ccr7, il17a, il10, and cd83. DisGeNet
[8] reported that cd4 is a therapeutic factor for arthritis infection, but we could not find it in the top-ranked paths with a depth of three.
There are 29 paths in the top-ranked path with a depth of four and 54 different relationships. Based on top-ranked paths with a depth of four, we found two types of diseases: HIV infections and malaria. HIV infection is an autoimmune disease that may have a higher risk in COVID-19. For malaria, there is a probability of misdiagnosis in COVID-19 and malaria
[13]. Compared to paths with a depth of three, paths with a depth of four involve more nodes but are less related to COVID-19 information.
2.2.2. STAT1 and Chloroquine
STAT1 is the primary transcription factor activated by interferons (IFNs) vital to normal immune responses, particularly viral, mycobacterial, and fungal pathogens
[14]. An innate immune response is a defense strategy that includes physical, chemical, and cellular level defenses. Type I IFNs are a critical component of this response. In COVID-19 cases caused by the SARS-CoV-2 N protein that inhibits the phosphorylation of STAT1 and STAT2, the conditions also suppress IFN signaling
[15]. Chloroquine, also known as Chlorochin and Aralen
[16], has been studied to treat and prevent COVID-19.
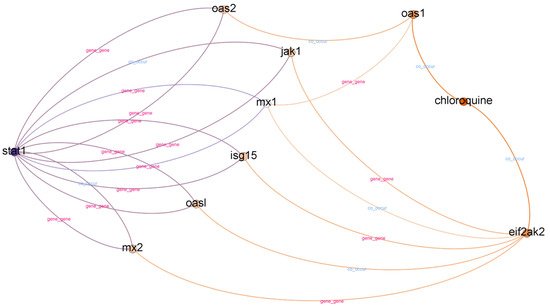
Chloroquine is an aminoquinoline primarily used to prevent and treat malaria in areas where it remains sensitive. Chloroquine is also vital as an anti-inflammatory agent in RA and lupus therapy. A subgraph from top-ranked paths with a depth of three is illustrated in Figure 4.
Figure 4. Subgraph from top-ranked paths with a depth of three in STAT1 and chloroquine case.
In contrast to IL-6 receptor–hydroxychloroquine, there are three paths with a depth of two in the STAT1–chloroquine case. We found six different relationships in paths with a depth of two. However, it is challenging to obtain information from paths with a depth of two. We found a “chloroquine–co_occur–weight loss” relationship in paths with a depth of two, but we could not find supporting evidence. The information obtained from paths with a depth of two is more related to mice.
In top-ranked paths with a depth of three, we found 31 paths and 15 different relationships. We found eight different gene nodes between the head and tail nodes in top-ranked paths with a depth of three. The gene nodes are MX1, MX2, ISG15, OAS1, OAS2, JAK1, OASL, and EIF2AK2. According to
[17], the MX1, ISG15, and OAS2 interferon-stimulated genes are potential candidates for drug targets in COVID-19 treatments. Furthermore, we found other evidence to support the relatedness of OAS1, JAK1, and OASL with COVID-19
[18][19][20]. However, we could not find supporting evidence for MX2 and EIF2AK2.
When we evaluated the top-ranked paths with a depth of four in the STAT1–chloroquine case, we found 51 paths and 56 different relationships. We found other genes in the top-ranked paths with a depth of four, such as USP18, A226V, SOCS, and LY96. USP18 is a differentially expressed gene (DGS) in COVID-19 cases
[19]. However, we could not find supporting evidence for hyperglycemiaA226V, SOCS, and LY96. Aside from co-occurring genes, we also found 15 diseases: dengue hemorrhagic fever (DHF), John Cunningham (JC) virus infection, dengue shock syndrome, alphavirus infections, hypoxia, pneumococcal pneumonia, pleural effusion, myalgia, asthma, empyema, hyperglycemia, bronchiectasis, arthralgia, bronchiolitis, and pneumonia.
In COVID-19 cases, there is a higher incidence of bilateral pneumonia and pleural effusion
[21]. The most common symptoms at diagnosis were coughs, myalgia, dyspnea, fever, and chills
[22]. In some cases, acute bronchiolitis with mucous membrane exfoliation, accumulation of bronchiolar secretions, and bronchiolar epithelial metaplasia occurred
[23]. A Spanish COVID-19 case series in Barcelona found that myalgia or arthralgia is a protective factor against ICU admission and death
[24]. Moreover, underlying lung disease, especially asthma, has recently been associated with a higher risk of hospitalization
[25].
As with the IL-6 receptor–hydroxychloroquine case, we can find more information using a higher depth (depth four) but obtain fewer significant paths than using depth three. However, results from STAT1–chloroquine are slightly different as chloroquine is also related to many other diseases. Therefore, in the STAT1-chloroquine case, there are more irrelevant nodes and information to COVID-19 extracted from top-ranked paths.
3. Conclusions
This study built a coronavirus KG by merging two existing datasets: AG and CORD-19. The combination of the two datasets enriches the KG with more entities. However, further analysis is needed to illustrate that those entities contribute to understanding the COVID-19 disease context. We analyzed our built KG using an ego network analysis for nodes, such as ACE, SARS, and PaO2. From the retrieved ego network, we can discover the high relatedness between those nodes and COVID-19.
We attempted pathfinding using a defined head and tail node to confirm KG usability for further knowledge discovery. We found that we could obtain paths with significant relationships using word-embedding and distance similarity between nodes. We also found that using a depth of three in both IL-6 receptor–hydroxychloroquine and STAT1–chloroquine cases resulted in more information related to COVID-19.
In the future, we plan to update this KG with more recent coronavirus publications. We also plan to include more related knowledge resources to enrich the graph. We will perform a further experiment in the COVID-19 domain query search for knowledge discovery using the built KG. We will explore more paths on scoring methods and missing link prediction.
 +1 point
+1 point