1000/1000
Hot
Most Recent
 +1 point
+1 point
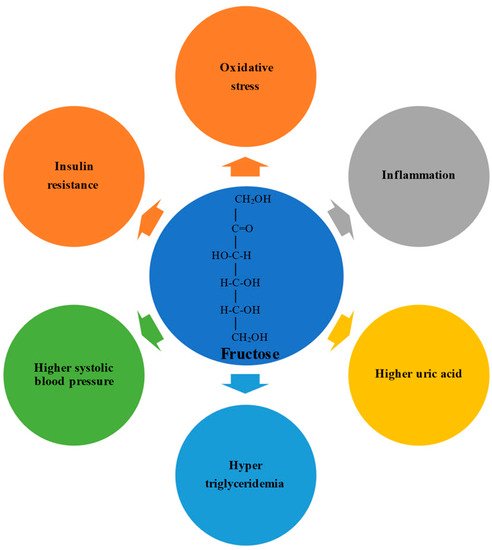
Fructose possesses an open-chain chemical conformation and is therefore much more reactive than glucose. Experimental studies have shown that a high fructose intake promotes oxidative stress, inflammation, higher serum uric acid levels, hypertriglyceridemia, higher systolic blood pressure, and insulin resistance(). In humans, the physiological impact depends on the formulation in which the fructose is consumed; consumption via solids and liquids differently affects microbiota composition, gut integrity, and liver toxicity.
Chronic diseases represent a major challenge in world health. Metabolic syndrome is a constellation of disturbances that includes dyslipidemia, type II diabetes, insulin resistance, visceral obesity, microalbuminuria, and hypertension [1][2]. The prevalence of metabolic syndrome is difficult to establish because there is no consensus on its definition [1], but estimations are 27.93% in North America, 27.65% in South America, 21.27% in Asia, 16.04% in Africa, and 10.47% in Europe [3], affecting a quarter of the world’s population [4]. The most important risk factors for developing metabolic syndrome are related to obesity, a complex disease associated with an imbalance between physical activity and calorie intake, and excessive consumption of fats and simple carbohydrates; the obesogenic environment also plays an important role [5].
Metabolic syndrome affects several organs, and it has been proposed to be a liver-centered condition [6]. Non-alcoholic fatty liver disease (NAFLD) is a term widely used to describe excessive fat infiltration in the liver in the absence of alcohol, autoimmune disorders, and viral hepatitis [7]. Moreover, NASH is a risk factor for liver cancer development [2][8][9][10][11][12]. HCC is the dominant form of primary liver cancer, which represents 75–90% of the total liver cancer burden [13][14].
A significant correlation between fructose intake and the degree of fibrosis has been found [15][16]. Its consumption has recently increased in many parts of the world because of the growing use of high-fructose corn syrup in beverages and processed food [17]. Studies on ancestral diets have shown that the average intake of fructose per capita was around 2 kg per year, while the current global average consumption of fructose per capita is 25 kg per year [18]. Furthermore, elevated consumption of fructose represents a great metabolic risk for not only obese but also lean individuals who have a high consumption of fructose-sweetened beverages
Fructose possesses an open-chain chemical conformation and is therefore much more reactive than glucose [19]. Experimental studies have shown that a high fructose intake promotes oxidative stress, inflammation, higher serum uric acid levels, hypertriglyceridemia, higher systolic blood pressure, and insulin resistance [20][21] (Figure 1). In humans, the physiological impact depends on the formulation in which the fructose is consumed; consumption via solids and liquids differently affects microbiota composition, gut integrity, and liver toxicity [22][23].
Sensory stimulation is the adaptive response to food intake through rapid physiological processes, and one of the most studied is the cephalic-phase insulin response. Additionally, eating fructose, in contrast to glucose consumption, leads to increased hunger and desire to eat because fructose decreases leptin and glucagon-like peptide 1 and increases ghrelin levels in the serum [24]. Ghrelin activates the neuronal activity of neuropeptide Y, increasing food intake, and glucagon-like peptide 1 inhibition causes a decrease in insulin secretion [25]. Regarding the hedonic value of fructose and the sum of all these events that affect appetite control, more studies are required to understand the role of fructose in the reward system.
The intestinal epithelium is the cell layer closest to the intestinal lumen and is composed by 70–80% of enterocytes, Paneth cells, goblet cells, and intestinal stem cells. Studies attribute the metabolic effects of fructose to enterocytes, cells specialized in absorption [26].
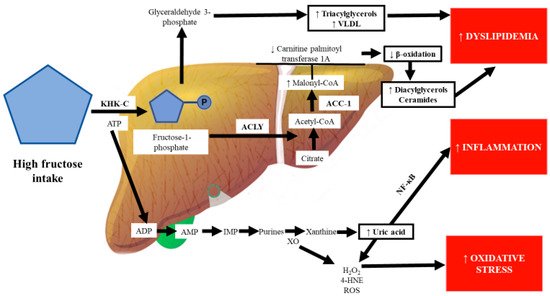
In humans, fructose is converted to glucose when the intake is moderate (≤1 g/kg of body weight), while high fructose consumption leads to the strong induction of Glut5 but not Glut2, thus increasing the fructose concentration and catabolism in the cytosol of intestinal epithelial cells [19][27][28][29]. In the cytoplasm of intestinal cells, the ketohexokinase (KHK) enzyme, also called fructokinase, which has a high affinity for fructose, phosphorylates fructose to fructose-1-phosphate, a toxic metabolite [30]. By contrast, KHK-C overexpression promotes intestinal fructose clearance and increases fructose-induced lipogenesis in the liver [30]. However, when the capacity for intestinal fructose clearance is exceeded, the increased activity of KHK-C exhausts adenosine triphosphate (ATP) and induces adenosine monophosphate deaminase activation, which results in marked ATP depletion, leading to the accumulation of adenosine monophosphate and uric acid production [31].
Uric acid, a weak organic acid end-product of purine catabolism in humans, is an antioxidant molecule that plays an essential role in the cardiac, vascular, and central nervous systems because it can neutralize pro-oxidant free radicals, such as hydroxyl radicals, hydrogen peroxide, and peroxynitrite [31]. Uric acid is produced in the liver and gut and excreted through the urine and feces [31]. According to Yun et al., the duodenum plays an important role in the synthesis and elimination of uric acid; one-third of the total uric acid is excreted through the gut [32]. Thirty percent of uric acid is excreted via ATP-binding cassette subfamily 2 (breast cancer resistance protein) on the luminal surface of the intestine, but an imbalance in its production or excretion can increase uric acid levels, favoring nicotinamide adenine dinucleotide (NADPH) oxidase (NOX) activation in the liver, acting as a damage-associated molecular pattern (DAMP) [33][34].
ChREBP is a transcription factor activated by a high-fructose diet, improving the KHK and Glut5 capacity for fructose absorption [35]. SREBP is a family of transcription factors consisting of three isoforms that regulate the homeostasis of lipids. In enterocytes, apolipoprotein induces the transcription of SREBP1c, which improves the stability of ApoB-48, the structural protein for chylomicrons, enhances microsomal triglyceride transfer protein, and augments lipogenesis [36]. This uncontrolled lipid metabolism and lower clearance of chylomicrons in the intestinal cells, together with uric acid overproduction, is responsible for increased cardiometabolic risk and leads to the development of NASH [35][37][38].
NASH models showed that cytochrome P450 2E1 activity is linked to increased intestinal inflammation during fructose consumption [39]. Furthermore, NASH patients have increased cytochrome P450-2E1 and inducible nitric oxide synthase, which cause the nitration of intestinal tight and adherent junction proteins [40]. The disruption of tight junction proteins and elevated apoptosis of enterocytes, evidenced by the upregulation of caspase 3 and p-JNK after fructose exposure, contributes to endoplasmic reticulum stress, the accumulation of unfolded or misfolded proteins, and the dysfunction of the epithelial barrier, which result in increased gut permeability, allowing lipopolysaccharides (LPS) to translocate from the gut lumen to the portal tract, triggering an inflammatory response in the liver [40]. In animal models that use fructose-rich diets, the intestinal absorption of Ca2+is decreased, and Ca2+receptors are depleted, which leads to decreased antioxidant defenses (GPx, catalase, superoxide dismutase, etc., are exhausted), and endoplasmic reticulum stress occurs [41].
Recent reports show that fructose consumption alters the gut microbiota and their bacterial metabolites, in a manner that promotes the development and progression of NASH [42]. As a consequence, increased blood ethanol concentrations and/or ethanol metabolites can alter the host’s metabolism, generate reactive oxygen species, and active inflammatory pathways, suggesting that microbiota that produce alcohol can have important effects on the evolution of NAFLD [43][44][45]. Moreover, gut dysbiosis triggered by excessive fructose intake leads to intestinal bacterial overgrowth, a strong decrease in microbial diversity, and increased translocation of bacterial products and cytotoxins, stimulating inflammatory pathways in experimental and human NAFLD The dysregulated microbiota, disruption of intestinal tight junction proteins, elevated uric acid production, and toxic bacterial metabolites accelerate NASH progression.
A diet rich in fructose induces the hepatic de novo synthesis of fatty acids and triglyceride accumulation Once fructose exceeds the intestinal clearance capacity, it is driven to the portal vein, where a fructosemic state strongly and quickly induces mechanisms involved in its overflow to the liver, which is the principal organ for fructose metabolism [6][21]. However, the mechanisms of the hepatic cell types (hepatocytes, hepatic stellate cells (HSCs), and Kupffer cells) that are involved in the metabolism of fructose consumed in large quantities are poorly understood [36]. In particular, chronic high fructose consumption induces the aldolase B enzyme, which breaks down fructose to dihydroxyacetone phosphate and D-glyceraldehyde.
Similar to in mice, KHK expression is elevated in obese patients with advanced liver disease compared to in obese subjects without fatty liver In KHK-knockout mice, ATP citrate lyase (ACLY), acetyl-CoA carboxylase (ACC)-1, and fatty acid synthase (FASN) are decreased by fructose administration [46]. ACLY is an enzyme that links carbohydrate to lipid metabolism by converting citrate to acetyl-CoA for fatty acid and cholesterol biosynthesis. ACC-1 coordinates the synthesis of fatty acids in the liver and generates a pool of malonyl-CoA used by FASN to generate palmitate [47].
Kupffer cells play a central role in liver damage induced by fructose. The elevated endotoxemia and oxidative stress produced by fructose intake promote hepatic Toll-like receptor (TLR)-4 activation. As previously mentioned, fructose causes gut-barrier deterioration through the disruption of tight-junction proteins. LPS and other bacterial toxins cross the gut barrier and bind to TLR-4 on the macrophages or Kupffer cells’ plasma membranes, which activates the proinflammatory signaling pathway, with a consequent increase in the expression of proinflammatory cytokines including tumor necrosis factor-alpha (TNF-α), interleukin (IL)-6, and IL-1beta (β)
Studies performed in mice models have shown that fructose triggers the infiltration/activation of macrophages/Kupffer cells, causing increased levels of ROS, and induces the necrosis of hepatocytes through TNF-α and IL-6 upregulation (90). Cytokine release causes hepatocyte death along with activation of HSCs and Kupffer cells. The NLRP3 inflammasome, upregulated by fructose overfeeding, is a sensor of danger signals, DAMPs, uric acid crystals, or derivatives that act like DAMP molecules and induce inflammation [48][49][50]. The activation of the NLRP3 inflammasome is a synchronized interaction between hepatocytes and Kupffer cells that results in dyslipidemia and lipid accumulation in hepatocytes [51].
Wree et al. found that NLRP3 inflammasome activation results in severe liver inflammation and fibrosis via the pyroptotic signaling pathway in hepatocytes [52]. Pyroptosis is a unique form of programed cell death where a plasma membrane pore formed by gasdermin D allows the release of the cellular content, leading to the upregulation of proinflammatory cytokines and profibrogenic factors such as IL-1β, connective tissue growth factor, and TGF-β, triggering the activation of HSCs, leading to the increased production and secretion of scar tissue proteins [52]; as a result, inflammation is exacerbated and liver fibrosis ensues [53][54]. Inflammasome activation by fructose could also be the result of increased Glut5 activity, which induces TXNIP to form the activated complex of ASC with NLRP3, consequently inducing dyslipidemia, hepatic inflammation, and lipid accumulation [55]. In addition, there is evidence indicating that TXNIP is upregulated in the liver by the master nutritional regulator ChREBP [56].
Keap1 acts as a sensor for oxidative stress, and under stress conditions, the sequestration complex dissociates, allowing Nrf2 to translocate to the nucleus, where it binds to the antioxidant response element and induces the expression of a battery of antioxidant genes [54]. Transcriptomic analysis reveals that the excessive consumption of fructose induces mechanisms that increase oxidative stress, such as aryl hydrocarbon receptor downregulation. The aryl hydrocarbon receptor modulates the expression of various biotransformation enzymes classified as phase I and II enzymes; this receptor also has crosstalk with NF- Therefore, fructose intake, which causes the downregulation of xenobiotic-metabolizing enzymes and Nrf2 transcription, also leads to the upregulation of NF-κB
By contrast, ACC-1 inhibition was associated with a decrease in hepatic de novo lipogenesis and insulin resistance and increased fatty acid β-oxidation [47]. FGF21 activates lipolysis and increases fatty acid oxidation in the liver through the activation of peroxisome proliferator-activated receptor alpha (PPAR-α). PPAR-α, which is mainly activated during the fasted state and regulates the metabolism of lipids and inflammation, is primarily found in hepatocytes, and fatty acids resulting from the metabolism of fructose are oxidized to produce acetyl-CoA by peroxisomes and mitochondria through PPAR-α [57]. By contrast, in FGF21-knockout mice, the activation of HSCs and fibrogenesis were increased, evidenced by increased levels of TGF-β, matrix metalloproteinases, and tissue inhibitors of metalloproteinases [58].
The respiratory chain of the mitochondria produces ROS, but ROS are decreased by antioxidant enzymes to prevent the deleterious effects of free radicals on important biological molecules. Fructose intake diminishes the antioxidant machinery of mitochondria, increasing oxidative stress, which causes the lipid peroxidation of polyunsaturated fatty acids, and allows the attack of free radicals on mitochondrial DNA; as a result, mitochondrial biogenesis is also affected [59]. Mitochondrial dysfunction results in low fatty acid oxidation, decreased hepatic ATP levels, and increased hepatic oxidative stress [60][61]. On the other hand, fructose oxidation also produces carbonyl compounds such as glycolaldehyde, a metabolite of glyceraldehyde, and glyoxal, the major product of glycolaldehyde oxidation, which is associated with cellular injury and dysfunction, including the inhibition of mitochondrial respiration and induction of mitochondrial permeability transition, leading to cell death [62][33][63].
Additionally, the consumption of fructose but not glucose increases apolipoprotein CIII through the ChREBP pathway, increasing triglyceride and low-density lipoprotein levels upon fructose metabolism, and represents a significant contributor to cardiometabolic risk [64][65]. These observations suggest that ChREBP plays an important role in the pathogenesis of NASH; however, the suggested protective role of ChREBP deserves further investigation [66].
The deleterious effects on lipid metabolism of excessive fructose consumption are fasting and postprandial hypertriglyceridemia, and increased hepatic synthesis of lipids, very-low-density lipoproteins (VLDLs), and cholesterol [64][65][67][68]. It has been shown that the elevated levels of plasma triacylglycerol during high fructose feeding may be due to the overproduction and impaired clearance of VLDL, and chronic oxidative stress potentiates the effects of high fructose on the export of newly synthesized VLDL [69]. Moreover, in humans diets high in fructose have been observed to reduce postprandial serum insulin concentration; therefore, there is less stimulation of lipoprotein lipase, which causes a greater accumulation of chylomicrons and VLDL because lipoprotein lipase is an enzyme that hydrolyzes triglycerides in plasma lipoproteins [70]. High fructose consumption induces the hepatic transcription of hepatocyte nuclear factor 1, which upregulates aldolase B and cholesterol esterification 2, triggering the assembly and secretion of VLDL, resulting in the overproduction of free fatty acids [71].
In hepatocytes, cytoplasmic Ca2+is an important regulator of lipid metabolism. A high fructose intake induces lipid accumulation, leading to protein kinase C phosphorylation, stressing the endoplasmic reticulum [72]. A prolonged endoplasmic reticulum stress response activates SREBP1c and leads to insulin resistance [73][74]. Calcium signaling is also important for liver regeneration, and increased intracellular calcium homeostasis is known to be involved in tumor initiation, progression, and metastasis; therefore, the alteration of calcium homeostasis by high fructose consumption could be an important mechanism in the development of cancer [75][76].
Furthermore, some reports indicate that fructose supplementation leads to insulin receptor downregulation because protein-tyrosine phosphatase 1B activity decreases the phosphorylation of the insulin receptor and induces protein phosphatase 2A, increasing SREBP1c, aggravating hepatic insulin resistance via intricate metabolic pathways [77]. Extensive reviews have been published on the lipogenic effect of fructose [35][60][78]; however, the deleterious effects of fructose in the liver go beyond the steatotic effect. Hepatic cholesterol accumulation is associated with inflammatory cell infiltration [79]. Dietary fructose induces strong SREBP1c activation, and the consequent palmitate production causes lipotoxicity in the endoplasmic reticulum; these events are the leading factors responsible for the greater Nrf2 inhibition and more intense hepatic inflammatory response driven by NLRP3 inflammasome activation [51][80].
Some authors have proposed “multiple parallel hit” theories to explain the development of the metabolic disease NAFLD, the first hit being the accumulation of fat in the liver (mainly triglycerides), followed by multifactorial processes that involve oxidative stress, inflammation, and hyperuricemia as the main factors [81][82]. DNA methylation is an epigenetic mechanism that decreases gene expression. Accumulating evidence suggests that excessive fructose intake drives epigenetic alterations, including the hypermethylation of the carnitine palmitoyl transferase 1A and PPAR-α genes [83][84]. Increased malonyl-CoA, which is synthesized by the enzyme acetyl-CoA carboxylase, inhibits carnitine palmitoyl transferase 1A, which is the rate-limiting step of the oxidation of lipids in the mitochondria, leading to the disruption of β-oxidation and accumulation of hepatic lipids, particularly fatty acids such as diacylglycerols and ceramides, which inhibit the insulin signaling pathway through protein kinase C activation and the inhibition of the protein kinase AKT, respectively [85][84] (Figure 2).
This scenario can be worsened because high-glycemic diets induce the conversion of glucose to fructose by the aldose reductase enzyme. Fructose can be endogenously synthesized in the body via the polyol pathway, a two-step conversion of glucose to fructose, which is relatively inactive under physiological conditions [86][87]. In addition, in high-glycemic diets, glucose is metabolized by fructose-3-phosphokinase to a highly reactive molecule, fructose-3-phosphate, causing the formation of advanced glycation end products, which can trigger inflammatory pathways through the activation of signaling pathways such as NF-κB and mitogen-activated protein kinases, aside from increasing lipogenesis and the disruption of β-oxidation, independently of caloric intake and weight gain [60][88][89]. On the other hand, fructose can be released from the liver to the systemic circulation and filtered and excreted by the kidneys, a decisive organ for fructose disposal, increasing metabolic abnormalities [90][91].
Chronic fructose consumption stimulates purine nucleotide turnover, which culminates in the synthesis of uric acid from xanthine by XO, leading to uric acid accumulation within hepatic cells [92]. 4-Hydroxynonenal, which is formed by the attack of ROS on biological membranes, induces XO, a key enzyme in purine and free radical metabolism; in turn, high activity of XO may further promote oxidative stress in the liver [93][94]. Increased systemic oxidative stress is recognized as an essential cause of elevated uric acid and inflammation [95]. Uric acid directly inhibits endothelial nitric oxide synthase; the impairment of nitric oxide synthesis decreases vascular smooth muscle relaxation and increases systolic blood pressure, leading to hypertension [96].
Oxidative stress and uric acid are amplifying actors that activate the nuclear factor of activated T cells, which plays a role in the regulation of inflammation and upregulates aldose reductase via the polyol pathway, leading to hepatic steatosis [87]. Oral and coworkers (2019) found a positive correlation between the degree of liver damage and uric acid concentration in non-obese and young patients with NAFLD, who had higher uric acid concentrations than the healthy control group [97]. Therefore, the activation of the NLRP3 inflammasome is associated with liver disease progression from simple fatty liver to NASH with inflammation and fibrosis [98]. These studies support the role of uric acid as a risk marker of liver damage via NLRP3 inflammasome activation; moreover, it represents a non-invasive marker and a possible predictor of NASH.
There is cumulative evidence that some miRNAs regulate several signaling pathways, leading to oxidative stress and inflammation in the liver. For example, in humans the elevation of miR-214 levels decreases glutathione reductase and cytochrome P450 activities; consequently, hepatic oxidative stress is augmented [99]. Mice with miRNA-29a overexpression show decreased DNA oxidative damage in an NAFLD model, suggesting its role in neutralizing oxidative stress [100]. MiRNA-149-5p is induced by uric acid in hepatocytes, causing lipid accumulation via the upregulation of FGF21, a protein implicated in lipid metabolism that is considered an anti-metabolic-syndrome hormone, therefore playing an important role in the prevention of NAFLD development [101].
In 1924, Otto Warburg described that cancer cells could obtain energy by fermenting glucose into lactate, and this is called the “Warburg effect” [102]. Importantly, several investigators have suggested that high fructose intake not only promotes cancer development in various tissues but also proposed that endogenously produced fructose in cancer cells could potentially stimulate cancer growth [102]. The key enzyme that stimulates endogenous fructose production is aldose reductase in the polyol pathway. Notably, fructose can be utilized by cancer cells as an energy source and, subsequently, for the synthesis of nucleic acids through the pentose phosphate pathway.
Plenty of evidence indicates that fructose and its metabolites play a significant role in the development of liver disease. Although direct extrapolation from animal findings to humans is not recommended, basic research has illuminated some of the cellular and molecular mechanisms that are involved in the deleterious effects of the overconsumption of fructose, including oxidative stress, inflammation, higher serum uric acid levels, hypertriglyceridemia, higher systolic blood pressure, insulin resistance, fibrosis, cirrhosis, and HCC. Free radical and uric acid overproduction induced by excessive fructose consumption also play pivotal roles in fatty liver, inflammation, fibrosis, and HCC progression through a variety of signaling pathways. However, more in-depth studies dealing with the involved molecular mechanisms of fructose-driven fibrogenesis are required to find new therapeutic targets for drug development to prevent hepatic fibrosis.
The alarming increase in metabolic syndrome and comorbidities can only be attenuated if the consumption of fructose, mainly in soft beverages, is significantly reduced worldwide. In addition, an active lifestyle incorporating the practice of sports seems to be useful for fighting the sedentarism associated with obesity. Patients suffering from hepatic maladies should be recommended to reduce fructose consumption to prevent aggravation of their condition because fructose may act as a conjoint pathological agent.





