Visceral leishmaniasis (VL) is one of the clinical forms of leishmaniasis, caused mainly by the intracellular protozoan Leishmania donovani or Leishmania infantum.
1. Introduction
Visceral leishmaniasis (VL) is one of the clinical forms of leishmaniasis, caused mainly by the intracellular protozoan
Leishmania donovani or
Leishmania infantum. Although there exist many experimental animal models for VL (e.g., rodents, dogs, monkeys, etc.), none of these models accurately reproduces what happens in humans (reviewed in
[1]). The Syrian golden hamster is considered to be the best experimental model to study VL (reviewed in
[1]), since it closely mimics the clinicopathologic features observed in naturally infected dogs and humans. However, the use of Syrian hamster models is limited due to the unavailability of some reagents
[2][3]. The murine animal model (mainly the BALB/c and C57BL/6 strains) has been extensively used in the immunological study of this disease. BALB/c is a susceptible strain that develops a life-long chronic infection which is not fatal to the host
[3]. Although experimental murine models of VL do not allow exact extrapolations with subclinical infection in humans, they have nonetheless been useful in identifying genes and predicting their functional roles in protective immune response (reviewed in
[1]). For these reasons, we decided to use BALB/c as the experimental model in this study. When the infection is reproduced in an experimental model, an immune response occurs in the main affected organs, liver and spleen. In the liver, the formation of cellular infiltrates or inflammatory granulomas occurs around the infected macrophages. Kupffer cells (KC) are key in this process, since these are the main tissue macrophages that cover hepatic sinusoids and are the main target of infection by strains of
Leishmania causing VL
[4][5].
The study of transcriptional changes at the tissue level represents a significant strategy to evaluate host–pathogen interactions and to elucidate the mechanism of pathogenesis of experimental VL. Many studies have focused on the transcriptomic analysis of immunological response in the spleen in
Leishmania-infected experimental hosts
[6][7][8][9], where many mediators were found to play a role in M1/M2 and Th1/Th2 responses, and also markers of T reg, Th17, and Tfh cells are involved (reviewed in
[10]). However, transcriptomic analysis regarding immunological response mechanisms in the liver (where infection develops shortly after inoculation) are scarce
[9]. Additionally, most studies in infected livers have been carried out after the acute phase when parasitic load is plateauing and infection control is underway
[9]. Interestingly, transcriptomic analysis of the immunological response to
L. donovani infection using blood, spleen, and liver tissue of BALB/c mice has shown a remarkably common signature with the upregulation of interferon responsive genes in all tissues
[9]. These sort of studies are useful for the identification of the pathways and immunological processes involved in infection, and have been used in the search for immunomodulatory strategies
[6][11][12]. One example of this in experimental VL is type I interferon signaling. Manipulation of this pathway was found to be an encouraging strategy for improving disease outcomes in VL patients
[13]. The in-depth analyses of the processes that drive early infection will provide a better insight into the immunological strategies that could control
L. infantum infection.
The methodology used in this study is based on high-throughput real-time qPCR for the amplification of 223 genes related to both the immune response and lipid and carbohydrate metabolism. This is a highly sensitive and specific technology designed to perform large-scale gene expression analyses, allowing the correction of experimental variations by normalization. High-throughput real-time qPCR presents one important advantage over the most widely used methodologies in transcriptomics (RNA-seq, microarrays, etc.), namely that the results do not require validation by qPCR. Gene expression is also well-correlated with protein expression for numerous markers (such as IL12, IL10, IFN-γ, TLR2-4, TNF-α) in murine models
[14]. In this large-scale analysis, three different parameters were integrated: parasite burden in liver tissue, the weight of the organ, and gene expression profile in the liver at different timepoints. This large dataset was streamlined using principal component analysis (PCA) in order to identify a particular gene signature for some timepoints based on the contribution of each gene’s expression. PCA analysis revealed strikingly different gene-expression patterns between timepoints, dependent on the molecular processes taking place during early infection.
2. Leishmania Infantum Infection Is Established in Liver 24 h Post Inoculation
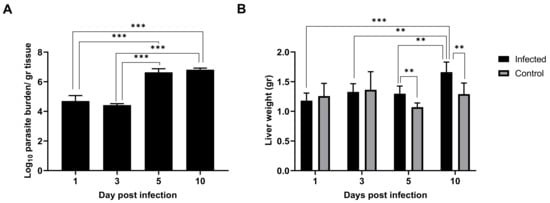
As early as 24 h post inoculation, high parasitic burden was detected in the liver tissue, indicating that the infection was already established in this organ (Figure 1A). This parasitic burden remained stable until three days p.i. At five days p.i., parasitic burden jumped two orders of magnitude and remained elevated subsequently. Hepatomegaly in the infected mice groups was not evident one and three days p.i., but small differences arose at five days p.i. and particularly at ten days p.i. (Figure 1B), which is in agreement with the leap in parasitic burden observed (Figure 1A). Given our results, it seems clear that although the liver was already colonized 24 h after L. infantum inoculation, clinical signs (such as patent inflammation) appeared a few days later in the infection in BALB/c mice.
Figure 1. Leishmania infantum infection is established in liver 24 h post inoculation: (A) Evolution of parasite burden in liver. Mice were inoculated intravenously (i.v.) with 1 × 106 promastigotes. Parasite load was determined 1-, 3-, 5- and 10-days post infection by limiting dilution assay and expressed as log10 of the average parasite load per gram of tissue. (B) Evolution of liver weight in infected (n = 24) vs. control mice (n = 24) over the course of infection. Error bars indicate Standard Error of Mean (SEM). One-way analysis of variance (ANOVA) was used to compare the differences in parasite burden between timepoints. Two-way ANOVA was used to compare the weight of liver between infected and control mice at each timepoint. Statistically significant differences are indicated (** p < 0.01, *** p < 0.001).
3. Multivariate Analysis Identified Particular Gene Expression Profiles at 1- and 10-Days Post Infection
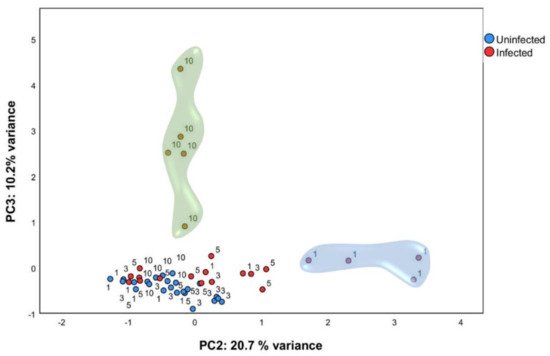
In order to identify gene expression profiles in this large dataset, PCA analysis was performed using normalized relative quantity (NRQ) of all infected (n = 24) and control (n = 24) mice at all timepoints (Figure 2). Four components were extracted with 60.84 % (cumulative) of the total variance explained. Principal component 2 (PC2) and principal component 3 (PC3) were used for subsequent analysis (Table 1). PCA analysis showed that, one day post infection, mice infected with L. infantum formed a subset (Figure 2, indicated by a blue cloud). This was confirmed by two-way analysis of variance ANOVA (p < 0.01), having the score of PC2 as the response variable and time post infection (1, 3, 5, and 10 days post infection) and condition (either infected or control) as factors. Therefore, as early as 24 h post-inoculation, infected mice showed a clearly divergent gene-expression profile driven by the expression of genes correlated to PC2 as a response to infection by L. infantum (Table 1).
Figure 2. Multivariate analysis identified particular gene expression profiles at one and ten days post infection. qPCR was carried out on liver tissue from control and L. infantum-infected mice and at 1, 3, 5, and 10 days post infection. Principal component analysis (PCA) plot. Principal components scores are shown on the X and Y axis, respectively, with the proportion of total variance related to that principal component (PC) indicated. Each dot represents a mouse in the corresponding day, and the color indicates the condition (red, infected; blue, control). The clouds highlight the group of ten days p.i. mice by PC3 (green cloud) and the group of one day p.i. mice by PC2 (blue cloud).
Table 1. Highest factor loadings of PC2 and PC3 from the Principal Component Analysis (PC2/PC3-correlated genes).
| PC2 Genes |
| Gene |
Factor Loading |
Gene |
Factor Loading |
Gene |
Factor Loading |
| Il18r1 |
0.742 |
Cd8a |
0.683 |
Tlr2 |
0.594 |
| Pdcd1lg2 (Pd-l2)e |
0.738 |
Cd4 |
0.682 |
Cd160 |
0.592 |
| P2rx7 (P2x7)a |
0.733 |
Ptgesa |
0.679 |
Irf1 |
0.591 |
| Bcl2 |
0.723 |
Il2rg |
0.678 |
Myd88 |
0.585 |
| Gata3 |
0.72 |
Ptger4a |
0.677 |
Pla2g4ab |
0.582 |
| Il16 |
0.719 |
Tnfrsf18 (Gitr)f |
0.668 |
Stat1 |
0.58 |
| Cd3e |
0.718 |
Il2rb |
0.653 |
Ccl2 |
0.58 |
| Il2ra (Cd25)f |
0.717 |
Tnfrsf4 (Ox40)f |
0.652 |
Il18bp |
0.577 |
| Lta |
0.713 |
Klrd1 (Cd94)c |
0.639 |
Irf5 |
0.573 |
| Il10ra |
0.713 |
Cd209dc |
0.634 |
Tlr4 |
0.572 |
| Cd69 |
0.712 |
Itgam |
0.633 |
Il6st |
0.572 |
| Akt3d |
0.707 |
Icam1 |
0.632 |
Ltb4r1 (Blt1)a |
0.571 |
| Cd28 |
0.706 |
Alox12a |
0.632 |
Il1b |
0.569 |
| Il7r (Cd127) |
0.704 |
Xcl1 |
0.622 |
Nfkb1 |
0.567 |
| Hpgds (Ptgds2)a |
0.704 |
Ucp2 |
0.621 |
Itgal |
0.567 |
| Mki67 (Ki67) |
0.702 |
Pdcd1lg1 (Pd-l1)e |
0.619 |
Cxcl9 |
0.567 |
| Ccr4 |
0.701 |
Cxcl10 |
0.614 |
Nfkb2 |
0.566 |
| Il9r |
0.697 |
Ccl22 |
0.614 |
Cxcl16 |
0.562 |
| Cxcr4 |
0.693 |
Ccr5 |
0.607 |
Cxcr6 |
0.552 |
| Ccl19 |
0.689 |
Irf7 |
0.605 |
Alox5a |
0.544 |
| Btla |
0.685 |
Itgb2 |
0.603 |
Rxrab |
−0.569 |
| Il18rap |
0.684 |
Cd68 |
0.6 |
|
|
| PC3 Genes |
| Gene |
Factor Loading |
Gene |
Factor Loading |
Gene |
Factor Loading |
| Il12rb1 |
0.943 |
Cxcr2 |
0.825 |
Cd83 |
0.63 |
| Il12rb2 |
0.941 |
Il27 |
0.803 |
Csf2 |
0.619 |
| Nos2 |
0.928 |
Cd80 |
0.782 |
Ccl4 |
0.614 |
| Ccr7 |
0.914 |
Irg1 (acod1) |
0.768 |
Havcr2 (Tim-3)e,f |
0.591 |
| Il12b |
0.884 |
Tgfb2 |
0.693 |
Icosl |
0.574 |
| Ifng |
0.867 |
Cxcr3 |
0.663 |
Ebi3 (Il27b) |
0.562 |
| Cxcr5 |
0.864 |
Ccl7 |
0.657 |
Il21r |
0.558 |
| Il23a |
0.842 |
Pdcd1 (Pd-1)e,f |
0.643 |
|
|
| Il21 |
0.839 |
Ptgs2a |
0.64 |
|
|
M1-associated transcripts are shown in bold; M2-associated transcripts are in italics; a Prostaglandin synthesis; b Lipid Metabolism; c C-type lectin receptors (CRLs); d MAPK signaling pathway; e Inhibitory markers; f Regulatory T cell function.
PCA analysis also showed that
L. infantum infected mice at ten days p.i. form another subset (
Figure 2, indicated by a green cloud). Using the score of PC3, we confirmed statistically significant differences between ten days p.i. mice and the control mice (
p < 0.001), showing that infected mice also had a clearly divergent gene-expression profile at this timepoint with the expression of genes included in PC3 (
Table 1). Additionally, unsupervised hierarchal clustering of the NRQ values was applied and, based on Euclidean distance and pairwise average linkage, was performed in order to visualize the relationships within this experimental dataset. Genes related to PC2 and PC3 are listed in
Table 1 and were classified depending on whether they are involved in specific biological processes or pathways. The list of genes correlated with PC2 include genes related to the immunological response triggered by infection, such as M1/M2 polarization
[8][15][16][17][18][19][20], but also genes involved in prostaglandin synthesis
[21][22][23], lipid metabolism
[24][25], MAPK signaling pathway
[26], and regulatory T cell function
[27] among others (
Table 1). This indicates a very early activation (24 h after infection) of diverse signals. In contrast, genes correlated with PC3 were not as numerous, and most of them are involved in Th1 response, suggesting the gene signature is tipping over to a more defined response at 10 days post infection. The most striking findings will be described below.
4. Multiple Processes Are Activated in L. infantum Infection in Livers of BALB/c Mice at 1-Day Post Inoculation
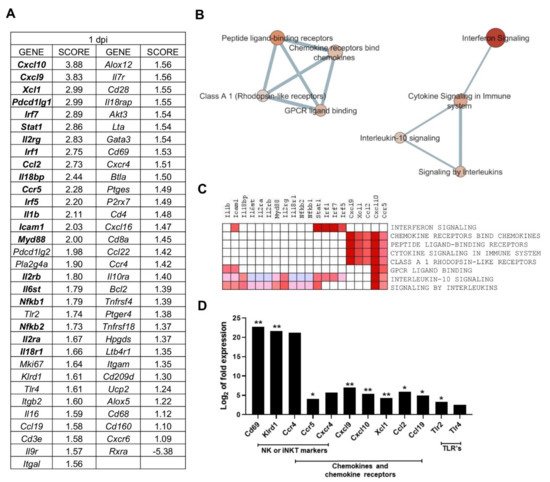
Gene Set Enrichment Analysis (GSEA) was applied to PC2-correlated genes (65 genes), to identify the most relevant gene-expression signatures in one day p.i. mice (Table 2). GSEA software ranked genes according to their differential expression between the groups compared (one day p.i. mice group vs. all other mice) (Figure 3A). Cxcl10 and Cxcl9 had the highest score in the ranked list (applying tTest), revealing the strong correlation between these genes expression and the gene signature observed for one day p.i. mice group (Figure 3A).
Figure 3. Multiple processes are activated in L. infantum infection in livers of BALB/c mice at 1-day post inoculation: (A) Ranked gene list generated in Gene Set Enrichment Analysis (GSEA) (using Principal Component 2-correlated genes to compare gene expression in 1 day p.i.-mice group vs. all other mice). Genes indicated in bold were involved in the core enrichment; (B) Enrichment Map with Principal Component 2 (PC2)-correlated genes. Parameters to filter these nodes/gene sets applied in Cytoscape: p-value < 0.05 and False Discovery Rate (FDR) < 0.25. Intensity of node color fill represents FDR value (more intense red color indicates lower FDR); node size corresponds to the Normalized Enrichment Score (NES) value. Thicker connection lines represent higher similarity coefficient between nodes; (C) Core enrichment of PC2-gene sets. The leading edge analysis was performed in GSEA with the gene sets enriched (with p-value < 0.05 and FDR < 0.25). The heat map shows the clustered genes in the leading edge subsets. Score values in the ranked gene list generated in GSEA are represented as colors, where the range of color (red, pink) shows the range of score values (high, moderate); (D) mRNA expression of genes at 1-day post infection expressed as Log2 of expression fold-change. Statistically significant differences between non- infected (control) and infected animals are indicated (* p < 0.05; ** p < 0.01).
Table 2. Gene sets upregulated in one day post infection-mice.
| Nº |
GENE SET NAME |
RATIO |
NES |
NOM p-Value |
FDR q-Value |
| 1 |
INTERFERON SIGNALING (REACTOME R-HSA-913531,2) |
5/149 |
2.050 |
0.000 |
0.039 |
| 2 |
CHEMOKINE RECEPTORS BIND CHEMOKINES (REACTOME DATABASE ID RELEASE 75 380108) |
10/49 |
1.782 |
0.013 |
0.199 |
| 3 |
PEPTIDE LIGAND-BINDING RECEPTORS (REACTOME R-HSA-375276,5) |
10/182 |
1.781 |
0.013 |
0.133 |
| 4 |
CYTOKINE SIGNALING IN IMMUNE SYSTEM (REACTOME DATABASE ID RELEASE 75 1280215) |
32/608 |
1.708 |
0.024 |
0.170 |
| 5 |
CLASS A 1 RHODOPSIN-LIKE RECEPTORS (REACTOME R-HSA-373076,8) |
12/307 |
1.629 |
0.036 |
0.227 |
| 6 |
GPCR LIGAND BINDING (REACTOME R-HSA-500792,3) |
12/420 |
1.629 |
0.035 |
0.190 |
| 7 |
INTERLEUKIN-10 SIGNALING (REACTOME R-HSA-6783783,3) |
6/42 |
1.584 |
0.040 |
0.212 |
| 8 |
SIGNALING BY INTERLEUKINS (REACTOME DATABASE ID RELEASE 75 449147) |
25/417 |
1.580 |
0.047 |
0.189 |
Results of the enrichment analysis performed in GSEA software, filtered by p-value < 0.05 and FDR < 0.25.
Enrichment analysis showed that eight gene sets were significantly enriched at FDR < 25% and nominal
p-value < 5% (
Table 2). The interferon signaling pathway was the most highly enriched pathway in one day p.i. mice, showing the highest NES value (normalized enrichment score, which was used to compare analysis results across gene sets
[28]) and lowest FDR. This gene set is linked to
Cytokine signaling in the immune system pathway, which in turn connects to signaling by interleukins and the interleukin-10 signaling pathway, according to the enrichment map (
Figure 3B). The observed enrichment is related to the upregulation of key genes of those pathways (
Figure 3C) and is characteristic of the one day p.i. mice group.
The second annotation with highest NES value was the Chemokine receptors bind chemokines pathway (
Table 2), forming a separate cluster connecting with several other annotations that involve processes of ligand-binding receptors (
Figure 3B). The overrepresentation of the “Chemokine receptors bind chemokines” Reactome pathway is in agreement with the upregulation of chemokines and chemokine receptors such as
Cxcl10, Cxcl9, Xcl1, Ccl2, and
Ccr5 (
Figure 3D). These genes were the core enrichment of this pathway, as described by GSEA, and contribute most to the enrichment result of this gene set (
Figure 3C). The upregulation of these genes is one of the earliest outcomes of
Leishmania infection described both in vitro and in vivo
[29][30][31][32][33]. This chemokine response at one day p.i. can be triggered by upstream signalling by toll-like receptors like
Tlr2 and
Tlr4 upregulated at this timepoint (
Figure 3D). In addition, some markers involved in toll-like receptor signaling and intracellular signaling (
Il1b, Icam1, Il18bp, Il6st, Il2ra, Il2rb, Myd88, Il2rg, Il18r1, Nfkb1, and
Nfkb2) were found in the core enrichment of the overrepresented “signaling by interleukins” annotation (
Figure 3C).
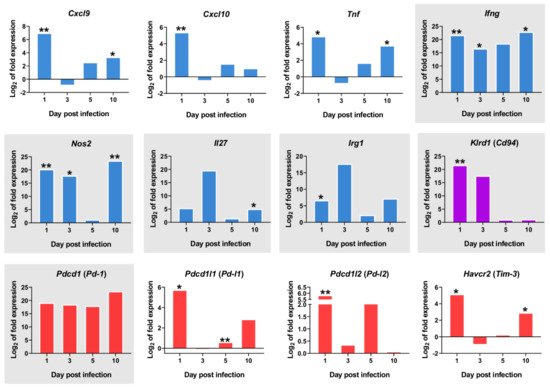
A number of other PC2-correlated genes show a clearly differential expression in the one day p.i.-mice group when compared to the other timepoints. Several of these correspond to genes expressed in natural killer (NK) cells or invariant natural killer T (iNKT) cells, such as Cd69, an iNKT cell marker, Klrd1 (alias Cd94, an NK cell marker), Ccr5, Ccr4 and Cxcr4 (Figure 3D). Despite the observed upregulation of M1 -associated transcripts at 1 day p.i. (Cxcl9, Cxcl10, Ifng, Tnf) (Figure 4), there was also strong upregulation of inhibitory ligands such as Pd-l1 (Pdcd1lg1) and Pd-l2 (Pdcd1lg2), Havcr2 (Tim-3), and Pd-1 (Pdcd1) (Figure 4).
Figure 4. mRNA expression of important markers overtime. The y-axis represents log2 of expression fold-change for each indicated gene, that is the ratio between the average gene expression in the infected group and non-infected-control mice. The x-axis represents time after infection 1, 3, 5, and 10 days post infection. M1-associated transcripts (blue bars), inhibitory markers (red bars), Klrd1 (Cd94) expression (purple bars). Graphics with gray background represent genes with Log2FC>1 in all timepoints. Statistically significant differences between non-infected (control) and infected animals are indicated (* p < 0.05; ** p < 0.01).
The overrepresentation of Interferon Signalling pathway (FDR = 0.039) with the upregulation of Stat1, Irf1, Irf7, Irf5, Icam1 (Figure 3C) and Chemokine receptors bind chemokines pathway (FDR = 0.199) with the upregulation of Cxcl10, Cxcl9, Xcl1, Ccl2, Ccr5 (Figure 3C) constitute the gene-expression signature 24 h after L. infantum infection in the livers of BALB/C mice.
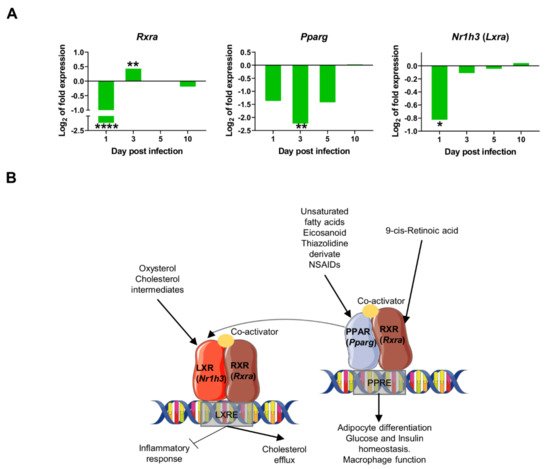
5. Downregulation of Transcriptional Factors Involved in Lipid Metabolism and Inflammation
In contrast to the positive correlation of most PC2 genes,
Rxra was the only gene negatively correlated with PC2 (
Table 1). Therefore at one day post infection (and only at this time of infection), the particular gene-expression signature observed in infected mice was dependent on the upregulation of PC2 genes, and downregulation of
Rxra. Rxra codes for Retinoid X Receptor Alpha (RXR-α), a nuclear receptor that mediates the biological effects of retinoids by binding, either as homodimer or heterodimer, to specific sequences in the promoters of target genes (
Figure 5B). RXR-α forms functional heterodimers with Peroxisome proliferator activated receptor (PPAR) proteins in a key pathway for lipid metabolism, adipocyte differentiation, and glucose and insulin homeostasis, among other functions (reviewed in
[34]). RXR-α also forms heterodimers with LXRA (Liver X Receptor-Alpha coded by
Nr1h3 gene) to bind LXR response elements (LXREs), regulating the expression of genes involved in macrophage cholesterol and fatty acid metabolism
[35]. Ligand activation of PPAR
γ has also been shown to induce LXR target genes and to promote cholesterol efflux in the presence of LXR ligands. Similarly, PPAR
γ and LXR ligands have been described to have anti-inflammatory effects, both
in vitro and
in vivo, through the inhibition of expression of genes involved in inflammation and innate immune response, such as iNOS, COX-2, TNF-
α, IL-6, IL1-
β among others
[36][37][38][39][40]. PPAR
γ activation downregulates proinflammatory cytokines in macrophages exposed to
L. infantum-Lipophosphoglycan (LPG), a surface molecule only found in promastigotes
[41]. In visceral and cutaneous leishmaniasis, several studies have demonstrated that the activation of macrophage PPAR
γ promotes disease progression, and its inhibition delays lesion development
[42][43][44] (reviewed in
[45]). In liver granulomas, it has been shown that sustained RXR-α /PPAR signalling is important for parasite growth and survival, and that infected macrophages are less susceptible to being polarized towards an M1 phenotype
[45] Similarly, pharmacological inhibition of RXR-α results in reduced parasite load in the spleen and liver
[25].
Figure 5. Downregulation of transcriptional factors involved in lipid metabolism and inflammation: (
A) mRNA expression of genes at 1, 3, 5, and 10-days post infection, expressed as Log
2 of expression fold-change. Statistically significant differences between non-infected (control) and infected animals are indicated (*
p < 0.05; **
p < 0.01; ****
p < 0.0001); (
B) In the macrophage nucleus, Nuclear Receptors (NR’s) Peroxisome proliferator-activated receptors (PPARs) and Liver X receptors (LXRs) bind to specific response elements (PPAR response element (PPRE) or LXR response element (LXRE)) in target genes as heterodimers with retinoid X receptors (RXRs). Activation of these NRs results in transrepression of proinflammatory genes (
Nos2, Ptgs2) but activation of cholesterol efflux, adipocyte differentiation, glucose, and insulin homeostasis, among others. This signaling is activated by several molecules like 9-cis-retinoic acid, unsaturated fatty acids, eicosanoids, oxysterol, and cholesterol intermediates. In our model, the downregulation of nuclear receptors (
Pparg, Rxra, Nr1h3) was observed at some timepoints shortly after infection, consistent with the overexpression of proinflammatory markers observed as early as one day post infection
[35][46][47].
Our data reveal the strong downregulation of several transcription factors at 1dpi: Rxra, Pparg, and Nr1h3 (Lxra) (Figure 5A), thereby suggesting an early inhibition of lipid metabolism routes like cholesterol efflux and fatty acid metabolism. This downregulation is transient since expression of all three markers return to control levels by 10 days post infection. The downregulation of those nuclear receptors probably contributes to the transcriptional induction of pro-inflammatory genes in the early innate immune response in the infection by L. infantum, such as Ifng, Nos2, and Tnf (Figure 4).
6. L. infantum Infection Induces Th1 Responses through Interleukin-12 Signaling at 10 Days Post Infection
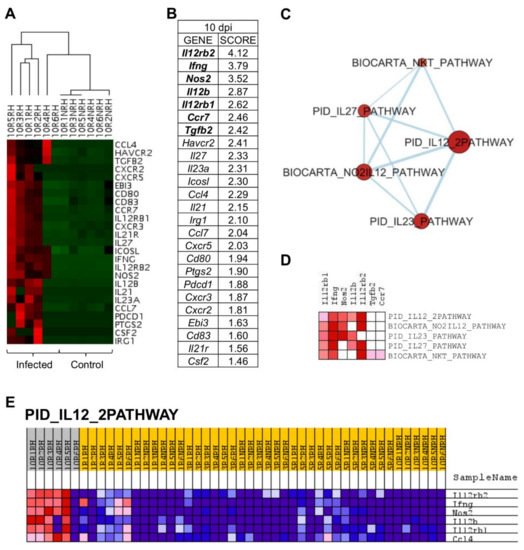
PCA allowed the discrimination of infected mice ten days post inoculation (Figure 2). This differentiation owes to the upregulation of some genes at that timepoint (Figure 6A), that are the genes correlated with PC3 in the principal component analysis (Table 1).
Figure 6. L. infantum infection induces Th1 responses through interleukin-12 signaling at 10 days post infection: (A) Unsupervised hierarchal clustering of 10 days p.i. and control mice including genes correlated with Principal Component 3 (PC3) was applied and based on Euclidean distance and pairwise average-linkage. Differential expression of selected genes in 10 days p.i.-mice (left side) and control mice (right side) is shown. Each mouse is identified with a respective code. Genes downregulated during infection are shown in green and upregulated genes are in red; (B) Ranked gene list generated in GSEA (using PC3-correlated genes to compare gene expression in 10 days p.i.-mice vs. all other mice). Genes indicated in bold were involved in the core enrichment; (C) Enrichment Map with PC3-correlated genes shows the relevance of IL-12 signaling pathway at 10 days p.i. Parameters to filter these nodes/gene sets applied in Cytoscape: p-value < 0.05 and FDR < 0.25. Intensity of node color fill represents FDR value (more intense red color indicates lower FDR); node size corresponds to the NES value. Thicker connection lines represent higher similarity coefficient between nodes; (D) Core enrichment of PC3-gene sets. The leading edge analysis was performed in GSEA with the gene sets enriched (with p-value < 0.05 and FDR < 0.25). The heatmap shows the clustered genes in the leading edge subsets. Score values in the ranked gene list generated in GSEA are represented as colors, where the range of color (red, pink) shows the range of score values (high, moderate); (E) PID_IL12_2PATHWAY heatmap. This gene set was upregulated in 10 days p.i.-mice (PC3 dataset). Upregulation of these genes in 10 days p.i.-mice (left side) and the rest of mice is shown. Each mouse is identified with a respective code. Genes are ordered by tTest ratio according to their differential expression. Normalized relative quantities (NRQ) are represented as colors, where the range of colors (red, pink, light blue, dark blue) shows the range of expression values (high, moderate, low, lowest).
Gene set enrichment analysis (GSEA) was applied to PC3-correlated genes (25 genes), revealing that Il12rb2, Ifng, Nos2, Il12b, Il12rb1, Ccr7, and Tgfb2 had the highest score in GSEA ranked gene list (Figure 6B) and, therefore, that they are key to the particular gene signature of 10 days p.i.-mice compared to the rest of mice in the experiment. An enrichment map was constructed using PC3-correlated genes to visualize the main functional categories (Figure 6C). Under these conditions, only five gene sets were upregulated by PC3 genes (Table 3). IL12-mediated signaling events (PID_IL12_2PATHWAY) was the annotation with the highest NES value and lowest FDR. Remarkably, genes involved in “PID_IL12_2PATHWAY” were upregulated in 10 days p.i.-mice group but not in the rest of the mice (Figure 6E), indicating their key role at this time of infection. The core enrichment of this pathway includes Il12rb2, Ifng, Nos2, Il12b, and Il12rb1 (Figure 6D). Other pathways upregulated in 10 days p.i.-mice group were related to IL23-mediated signaling events (PID_IL23_PATHWAY) with the upregulation of Ifng, Il12rb1, Il12b, Nos2, and Il23a. Additionally, IL27-mediated signaling pathway (PID_IL27_PATHWAY) was overrepresented, including Il12rb2, Ifng, Il12b, Il12rb1, Il27, and Ebi3. All these genes were correlated with PC3.
Table 3. Gene sets upregulated in 10 days post infection-mice.
| Nº |
GENE SET NAME |
RATIO |
NES |
NOM p-Value |
FDR q-Value |
| 1 |
PID_IL12_2PATHWAY (MSIGDB_C2 PID_IL12_2PATHWAY) |
6/59 |
2.188 |
0.000 |
0.003 |
| 2 |
BIOCARTA_NO2IL12_PATHWAY (MSIGDB_C2 BIOCARTA_NO2IL12_PATHWAY) |
5/15 |
1.965 |
0.003 |
0.013 |
| 3 |
PID_IL23_PATHWAY (MSIGDB_C2 PID_IL23_PATHWAY) |
5/36 |
1.879 |
0.006 |
0.016 |
| 4 |
PID_IL27_PATHWAY (MSIGDB_C2 PID_IL27_PATHWAY) |
6/25 |
1.788 |
0.012 |
0.024 |
| 5 |
BIOCARTA_NKT_PATHWAY (MSIGDB_C2 BIOCARTA_NKT_PATHWAY) |
8/25 |
1.642 |
0.035 |
0.052 |
Results of the enrichment analysis performed in GSEA software, filtered by p-value < 0.05 and FDR < 0.25.
Two pathways related to NK cells were overrepresented at this timepoint, due to the upregulation of characteristic markers: NO2-dependent IL-12 pathway in NK cells (BIOCARTA_NO2IL12_PATHWAY) (Il12rb2, Ifng, Nos2, Il12rb1, Cxcr3) and selective expression of chemokine receptors during T-cell polarization (BIOCARTA_NKT_PATHWAY) (Il12rb2, Ifng, Il12rb1, Ccr7, Tgfb2, Cxcr3, Ccl4, Csf2) (Figure 6A). The upregulation of markers involved in these pathways shows that macrophage activation is occurring at 10 days p.i. and the differentiation of CD4+ T cells is being promoted to the Th1 subset. Strikingly, the upregulation of inhibitory markers observed at one day p.i. is no longer evident at 10 days p.i. in livers of BALB/c infected mice (Figure 4). Among inhibitory markers, Havcr2 (Tim-3) was the only one showing statistically significant upregulation at 10 days p.i. (p = 0.022).
In conclusion, the gene-expression signature predominant in livers of BALB/c mice 10 days after infection with L. infantum is characterized by the overrepresentation of IL12-mediated signaling events (FDR=0.003) with the strong upregulation of Il12rb2, Ifng, Nos2, Il12rb1, and Il12b.
 +1 point
+1 point