1000/1000
Hot
Most Recent
 +1 point
+1 point
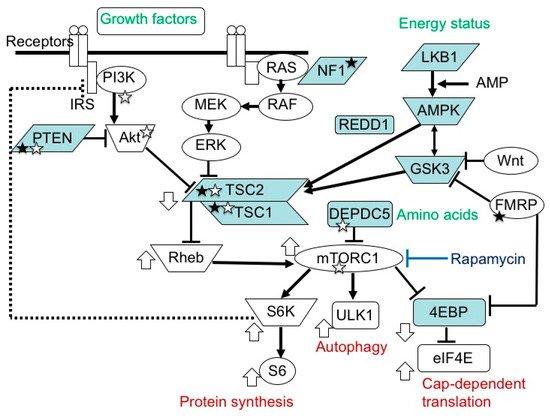
The mammalian target of the rapamycin (mTOR) system regulates various cellular functions, such as growth, proliferation, metabolism and survival/death. In systemic organs, it is critically involved in multiple processes, including neurogenesis, nutrition and immunity. In the brain, its roles are essential in the cerebral cortical development, synaptic functions, and brain activities such as learning, cognition and social functions.
| Organ | Hamartias | Hamartomas |
|---|---|---|
| Skin | Hypomelanotic macules Confetti skin lesions |
Facial angiofibromas Fibrous cephalic plaque Ungual fibromas Shagreen patch |
| Brain | Cortical tuber | SEGA Subependymal nodules |
| Eye | Retinal achromic patch | Retinal hamartoma |
| Mouth | Gingival fibromas | Dental enamel pits |
| Lung | Pulmonary LAM MMPH |
|
| Heart | Cardiac rhabdomyoma | |
| Arteries | Wall defects/Aneurysm | Renal AML |
| Kidney | Renal cysts | |
| Bone | Bone cysts |
| Lesion/Symptom | Pathology | Age of Occurrence or Worsening | Department in Charge | Note |
|---|---|---|---|---|
| Cardiac rhabdomyoma | Tumor | Fetal–neonatal period | Pediatric cardiology | Spontaneous regression in infancy |
| Cortical tuber | Dysplasia | Fetal–neonatal period/Infancy | Pediatric neurology | Focus of epileptic seizures |
| Hypopigmented macule | Dysplasia | Fetal–neonatal period/Infancy | Dermatology | |
| Epilepsy | Brain dysfunction | Infancy/Childhood | Pediatric neurology | Intractable in many cases |
| ID/ASD | Brain dysfunction | Infancy/Childhood | Pediatric neurology/Psychiatry | |
| Retinal hamartoma | Tumor | Infancy/Childhood | Ophthalmology | |
| Renal cyst | Dysplasia | Infancy/Childhood | Pediatrics | |
| SEGA | Tumor | Childhood/Adolescence | Neurosurgery | Hydrocephalus, potentially fatal |
| Facial angiofibroma | Tumor | Childhood/Adolescence | Dermatology | |
| TAND | Brain dysfunction | Childhood/Adolescence | Pediatrics/Psychiatry | |
| Renal AML | Tumor | Childhood/Adolescence/Adulthood | Pediatrics/Urology | Hemorrhage, potentially fatal |
| Pulmonary LAM | Tumor | Adolescence/Adulthood | Pulmonology | Predominantly affecting women, potentially fatal |