1000/1000
Hot
Most Recent
 +1 point
+1 point
It has been proposed that a “common core” of pathologic pathways exists for the large family of amyloid-associated neurodegenerations, including Alzheimer’s, Parkinson’s, type II diabetes and Creutzfeldt–Jacob’s Disease. Aggregates of the involved proteins, independently from their primary sequence, induced neuron membrane permeabilization able to trigger an abnormal Ca2+ influx leading to synaptotoxicity, resulting in reduced expression of synaptic proteins and impaired synaptic transmission. Emerging evidence is now focusing on low-molecular-weight prefibrillar oligomers (PFOs), which mimic bacterial pore-forming toxins that form well-ordered oligomeric membrane-spanning pores. At the same time, the neuron membrane composition and its chemical microenvironment seem to play a pivotal role. However, up to now the existence of a specific “common structure” of the toxic aggregate, and a “common mechanism” by which it induces neuronal damage, synaptotoxicity and impaired synaptic transmission, is still an open hypothesis. In this review, we gathered information concerning this hypothesis, focusing on the proteins linked to several amyloid diseases. We noted commonalities in their structure and membrane activity, and their ability to induce Ca2+ influx, neurotoxicity, synaptotoxicity and impaired synaptic transmission.
Amyloid proteins are a large family of proteins with the common tendency to aggregate through a process that, triggered by a misfolding event, involves first a slow and thermodynamically unfavorable nucleation phase followed by a rapid elongation phase (seeding-nucleation model) leading to the formation of mature fibers (MFs) [1][2]. The misfolding process can involve practically all proteins and has therefore been defined as their “dark side” [3].
Several highly diffused or rare human diseases are linked to the formation of amyloid aggregates through the misfolding process of the involved proteins. The most famous include amyloid-β (Aβ), α-synuclein (α-syn), amylin (hIAPP) and prion (Pr), which are linked to Alzheimer’s disease (AD), Parkinson’s disease (PD), type II diabetes and Creutzfeldt–Jacob’s Disease (C–JD), respectively [1][4]. Chiti and Dobson took stock of this topic in a recent, important and exhaustive review [1].
During the common aggregation process, which occurs both in vitro and in vivo, low-molecular-weight prefibrillar oligomers (PFOs) appear before MFs, rich in cross-β-sheets. PFOs can further aggregate with each other to form annular or linear protofibrils (APFs or LPFs) and, finally, MFs. Up to now, PFOs have been considered the most toxic species, responsible for cellular damage and subsequent amyloid toxicity [1][5][6].
A correlation between PFOs and a group of cognitive disorders involving the central nervous system (CNS) and known as “neurodegenerative diseases” has been demonstrated [7][8][9]. Although the symptomatology and epidemiology of these diseases have been extensively characterized [10], the molecular mechanism by which oligomeric aggregates induce neurotoxicity and cell death has not been fully elucidated. This is mainly due to the rapid rate and heterogeneous nature of the aggregation process, which amplifies the difficulty scientists have in identifying the amyloid aggregates responsible for toxicity [11][12]. As recently pointed out by Benilova et al., the tendency of proteins, such as Aβ, to rapidly aggregate during experiments leads to a difficult and uncertain identification of the structure responsible for a well-defined biologic effect [11]. For this reason, amyloid experimental models are also used, i.e., proteins, which although not associated with any pathology, undergo an aggregation process and exert typical neurotoxic effects both in vitro and in vivo.
Notably, several studies of both amyloid proteins and amyloid models have suggested that oligomers, regardless of the nature of the protein from which they derive, share common structure and mechanism of action [8][12][13][14]. It has been reported that a common feature of all neurodegenerative diseases is synaptotoxicity, resulting in reduced expression of synaptic proteins and impaired synaptic transmission by neurotoxic oligomeric aggregates [15][16][17][18]. Recently, Soto and Pritzkow provided a critical discussion on the role of protein misfolding and aggregation in neurodegeneration. They highlighted commonalities and differences between distinct protein aggregates and discussed evidence supporting the hypothesis that misfolded aggregates may be transmissible by the prion principle following a “cross seeding” behavior [19].
To further investigate the intriguing hypothesis of the existence of common structure and mechanism of action in the amyloid neurodegenerations, in the present review we will focus our attention on commonalities between i) structural features of the toxic PFOs of amyloid proteins and models and ii) functional effects PFO-induced on synaptic function and transmission, considered as the main molecular and electrophysiological mechanisms responsible for neuronal dysfunction related to cognitive impairment.
As argued by Glabe in 2006, soluble spherical aggregates of about 3–10 nm in diameter have been observed for many types of amyloid proteins by several microscopy techniques, and have been called micelles, PFOs, protofibrils and Aβ-derived diffusible ligands (ADDLs) [13].
Our interest will be focused on PFOs, which are the intermediate species formed during the aggregation process and are considered the most toxic species. In general, PFOs are defined as neurotoxic and soluble low-molecular-weight aggregates, spherical shaped with diameters in the order of nanometer, mainly in random configurations, different from the β-sheet configuration of amyloid fibers.
In a pivotal paper in 2003, Kayed et al. reported the production of an antibody that specifically recognizes micellar Aβ and not soluble, low-molecular-weight aggregates (low-MW) or fibrils. Notably, this antibody also specifically recognizes soluble oligomers among all other types of amyloidogenic proteins and peptides examined, including α-syn, hIAPP, polyglutamine, lysozyme, human insulin and PrP(106–126) [8]. Most important, this antibody was able to inhibit toxicity mediated by all other types of soluble oligomers. Thus, the authors concluded that the soluble oligomeric forms of all tested amyloids share a common structure that can mediate toxicity by a common mechanism.
Common features of cytotoxic amyloid species have been also described in 2010 by Bolognesi et al. for several proteins, such as wild-type Aβ1-42, the I59T variant of human lysozyme and an SH3 domain. Their results suggest a model in which the exposure of the hydrophobic surfaces, as a result of the aggregation of misfolded species, is a crucial and common feature of these pathogenic species [20]. These data strongly support the hypothesis that the exposure of hydrophobic patches is a key feature of the toxicity of extracellular oligomeric species. In the so-called “coalescence and reorganization” model, the generation of a stable hydrogen-bonded core drives the exposure of the hydrophobic residues that were buried in the initial collapse [21][22].
It has been suggested that the different types of oligomers, as they present similar structural features, could share a common mechanism of neurotoxicity. This neurotoxic action could be expressed through the deregulation of Ca2+ homeostasis and cell death [8][23]. It has been also reported that the lipid composition of membranes may modulate the cellular response to the toxic action of amyloid aggregates, thus explaining the different vulnerability of different cell types to amyloid species [24]. In particular, the importance of GM1, a major component of “lipid-rafts”, in the cellular membranes as a mediator of amyloid toxicity has been widely documented [25][26][27][28].
Kayed and Lasagna-Reeves, in 2013, reviewed the molecular mechanisms proposed for amyloid oligomer toxicity. Two kinds of mechanisms were proposed: “receptor-mediated” and “cellular membrane”. In the first, oligomers disrupt Ca2+ homeostasis, interacting with several cellular receptors, while in the second, forming ionic channels in the membrane [9]. In any case, the intracellular [Ca2+] rise seems to play a fundamental role in both paradigms (Figure 1).
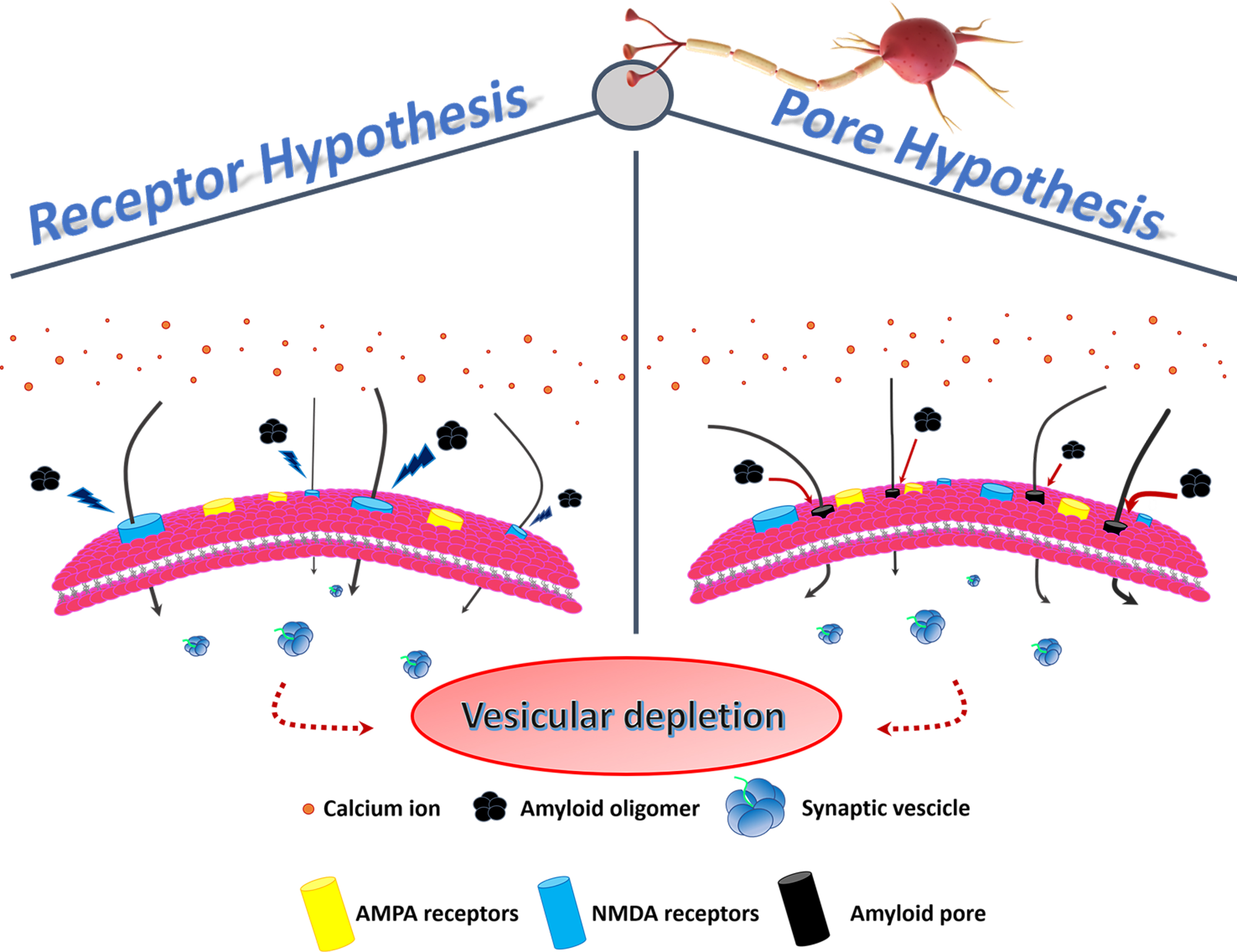
Figure 1. Membrane permeability and vesicular depletion. In the physiological conditon, membrane receptors regulate the entry of Ca2+ into the cell, ensuring the preservation of the pool of vesicles containing the neurotransmitter. In the pathological condition, characterized by the unbalance of the Ca2+ influx, two hypotheses have been formulated to explain the altered membrane permeability: in the first, PFOs directly activate NMDA receptors (receptor hypothesis), while in the second, PFOs form pores (continuous red arrows) in the membrane (pore hypothesis). Increased Ca2+ influx (black arrows) results in vesicular depletion (dashed red arrows) and consequent cell death.
Concerning “receptor-mediated” toxicity, several receptors have been considered to play a key role such as nerve growth factor or the N-methyl-D-aspartate (NMDA) receptors, which are a subtype of ionotropic glutamate receptors responsible for Ca2+ regulation [18][26][29][30][31][32][33]. However, in their review, Kayed and Lasagna-Reeves concluded that a major unknown was the identity of the receptor that bind oligomers and mediates neuronal dysfunction, and some studies are contradictory [9].
Amyloid proteins, such as PrP, Aβ, amylin (islet amyloid polypeptide—IAPP) and calcitonin (CT), interact with membranes, inducing damage via the formation of ion channels, the so-called “amyloid pore hypothesis” [34]. In particular, Kayed et al., in 2004 showed that only soluble oligomers, and not MFs, from several types of amyloids (Aβ, α-syn, IAPP, polyglutamine and PrP) specifically increased the membrane conductance of planar lipid bilayers, regardless of the protein sequence [23] and, in 2005, Quist et al. directly visualized by atomic force microscopy (AFM) typical amyloid pores for a group of six amyloid proteins [35]. The “amyloid pore hypothesis” was supported by Kagan et al., in 2004, who reported on the physiologic effects induced by this phenomenon, including Ca2+ dysregulation, membrane depolarization, mitochondrial dysfunction, inhibition of long-term potentiation (LTP) and cytotoxicity [36]. Finally, in 2019, Yasumoto et al., in an important and extensive paper, clearly demonstrated that Aβ oligomers disturbed membrane integrity, concluding that “membrane pore formation may also impair cellular and synaptic functions and is consistent with the observed loss of membrane integrity demonstrated by MTT, LDH, and calcein/ethidium homodimer-1 assay, [Ca2+] elevation, resting membrane potential increase input resistance decrease and LTP impairment”[37].
Soluble amyloid oligomers have a neurotoxic effect even when formed from proteins not directly related to neurodegenerative diseases, used as amyloid models [23][38][39].
In 2019, using salmon calcitonin (sCT) as an amyloid model due to its slow aggregation rate, allowing us to prepare stable samples without photochemical cross-linking, we tested the effects of native sCT PFO-enriched solutions in primary neurons and mice brain slices in terms of Ca2+ influx, cellular viability, LTP impairment, post-synaptic densities and synaptophysin expression. The results indicate that PFO-enriched solutions induced abnormal Ca2+ influx, which could only in part be ascribed to the NMDAR activation. Thus, we proposed an innovative “unified” neurotoxicity mechanism where both paradigms coexist, where the membrane permeabilization per se is not able to induce neurotoxicity but triggers an abnormal activation of the NMDAR [18]. Notably, in this paper, we obtained, by several techniques, experimental results very similar to those published by Yasumoto et al., for Aβ [37]. This similarity prompts the intriguing hypothesis in the open debate about the existence of a “common mechanism” in the pathogenesis of amyloid neurodegenerations.
We summarize the main information in Table 1, focusing on the principal synaptic proteins involved in amyloid toxicity and the effects caused by their altered expression.
The pathogenesis of amyloid diseases could be explained by a loss of plasticity that can negatively affect dendritic branching, synaptic remodeling, LTP, axonal sprouting and neurite extension, as well as synaptogenesis and neurogenesis processes [52][53][54][55]. Indeed, plasticity, the process by which synapses modulate their strength and form new neuronal connections, is known to play an essential role in response to injury and disease [56]. Moreover, most of the scientific evidence in the literature agrees that the synaptic plasticity loss is induced by PFOs [15][16][17][18][57][58]. For this reason, electrophysiological recordings of excitatory field potentials could represent an excellent strategy to assess the synaptotoxic effects of amyloid oligomeric aggregates.
|
Amyloid Oligomers |
Proteins Involved |
Synaptic Effects |
Neurodegenerative Disease |
References |
|
Aβ |
Synaptophysin |
Vesicular depletion |
AD |
[40] |
|
PSD-95 |
Decrease in dendritic spine density |
|||
|
MAP2 |
Dendritic tree reduction |
[41] |
||
|
Tau |
Synaptogyrin-3 |
Impair presynaptic function |
AD and tauopathies |
[42] |
|
Synaptophysin Septina-11 |
Impaired presynaptic density and neuronal trafficking deficit |
[43] |
||
|
α-syn |
SV2 SNAP25 |
Alteration of both neurotransmitter release and vesicle–membrane fusion |
PD |
[44] |
|
HTT |
Complexin II Synaptobrevin 2 |
Altered neurotransmitter release |
HD |
[45] |
|
SNAP25 Rhabphilin 3a |
Vesicle–membrane fusion and vesicle recycling deficit |
[46] |
||
|
Synaptophysin MAP2 |
Vesicular depletion and reduction in dendritic spine density |
[47] |
||
|
PrP |
GluR2 subunit of AMPAR |
Increased permeability to Ca2+ |
Prion’s disease |
[48] |
|
Synaptophysin |
Vesicular depletion |
[49] |
||
|
sCT |
Synaptophysin MAP2 |
Vesicular depletion and dendritic tree alteration |
No neurodegenerative disease |
[50] |
|
PSD95 |
Decrease in dendritic spine density |
[51] |
Amyloid-associated neurodegenerative diseases are characterized by intra- or extracellular accumulation of specific proteins in the CNS. Although these proteins differ in their primary sequence, they all share the tendency to adopt an incorrect conformation and the propensity to aggregate, starting from the formation of soluble low-molecular-weight PFOs to insoluble aggregates such as MFs. It has been proposed that PFOs, considered the most neurotoxic of all aggregates, share a “common structure” and a “common mechanism” by which they induce neuronal damage and death.
Concerning the PFOs some commonalities can be noted. Firstly, they are small globules ranging in diameter from 5 to 10 nm, formed by low‐molecular‐weight oligomers (tetramers - dodecamers) with a morphology that maximizes the surface/volume ratio, favoring its interaction with the cellular membranes. Their structure is generally metastable, with many proteins in the random configuration, and a more ordered but not‐crystalline core. In this arrangement, the oligomer surface is made of flexible and disordered domains that expose the hydrophilic protein parts to the solvent, together with stiff and more ordered hydrophobic patches only partially accessible to the solvent. The electrostatic interaction between the flexible and hydrophilic protein parts of the PFOs and the charged parts of the “lipid-rafts” in the membrane drives the binding to the target neurons. Notably, disordered aggregates lacking the core, or totally ordered aggregates such as spherical protofibrils, were unable to induce detrimental effects.
Concerning the PFOs’ neurotoxicity mechanisms, a common core of pathologic pathways exists based on the cellular membrane permeabilization and the subsequent abnormal Ca2+ influx, induced by aggregates of the involved proteins. It is generally accepted that PFOs form non-selective permeable “amyloid pore” and that “lipid‐rafts” play a special role. However, this process is not able to induce “per se” neurotoxicity and the involvement of ionotropic glutamate receptor NMDA, is necessary. The increase in the intracellular Ca2+ concentration is the key event that triggers the processes leading to cell death. The reserve pool of synaptic vesicles containing the neurotransmitter is depleted with the impairment of post‐synaptic structures, as evidenced by the reduced expression of proteins typical of dendritic spines.
This generalized scenario (Figure 2) leads to the functional effects observed in vitro with the impairment of the synaptic functions, and in vivo with the progressive cognitive deterioration.
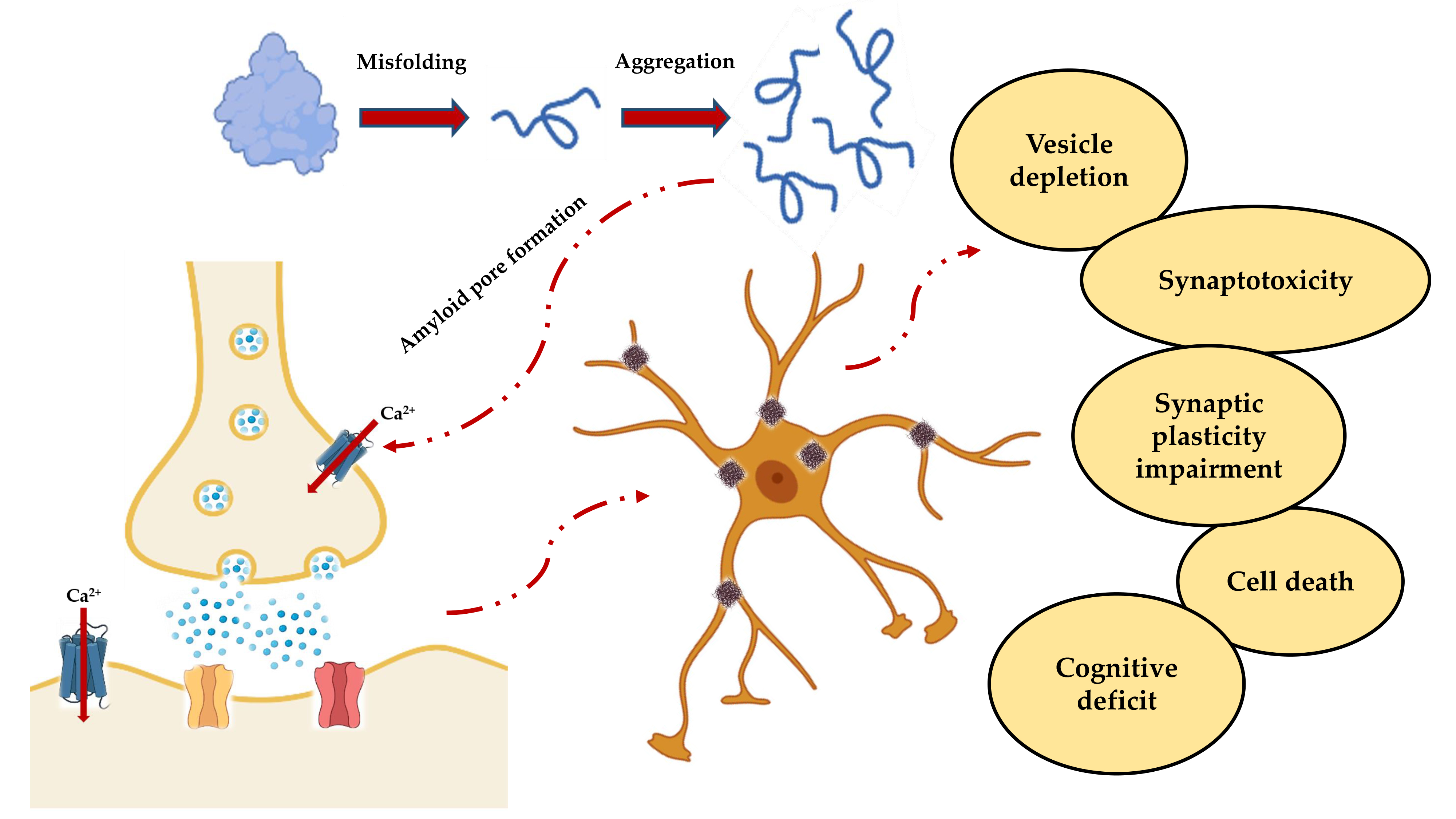
Figure 2. Schematic representation of the events leading to neuronal death and subsequent cognitive impairment. Misfolding and aggregation lead to the formation of PFOs, which not only result in the formation of insoluble fibers that are deposited in the nerve tissue, but also form amyloid pores in the synaptic membrane. The resulting deregulation of the Ca2+ homeostasis is responsible for the cascade of events that include vesicular depletion, synaptotoxicity, impaired synaptic plasticity and, ultimately, cell death and cognitive decline.





