“Free radical theory” of aging by Harman et al. propose that ROS production and accumulation, which was mainly produced by the disturbed mitochondrial function during aging, causes DNA mutations and degenerative diseases
[34]. However, it is becoming clear that small amount of ROS act as signaling molecules in the cells and they elicit health-promoting effects. Reduced glucose metabolism or impaired insulin/IGF signaling alters mitochondrial metabolism to extend life span in various model organisms
[3]. MtROS acts as a signaling molecule in these setting to activate beneficial retrograde signaling that regulates mitochondrial dynamics, quality control, proteostasis, biogenesis, and the cellular defense system. In the field of toxicology or radiation medicine, hormesis refers to the preconditioning events in which a subtoxic dose of a substance provokes an endogenous cytoprotective response and confers resistance against later higher amounts of the same toxicants. Patrick Tapia proposed the concept of “mitohormesis” such that weak mitochondrial stress mediates beneficial outcomes of calorie restriction, intermittent fasting, exercise, and dietary phytochemicals with accompanying stoichiometric amount of ROS being essential for the response
[35]. The theory was supported by following studies and now multiple pathways are known to mediate the mitohormetic response
[3][36]. Mitochondrial stress accompanies not only ROS production and ATP decline, but also accumulation of unfolded protein, decrease in Ca
2+ buffering, and alteration in metabolite of TCA cycle, OxPhos, fatty acid oxidation, etc.
[37]. Ca
2+ release from mitochondria in muscle during exercise stimulates mitochondrial respiration and biogenesis via calmodulin-dependent protein kinase and calcineurin
[38]. An increase in the NAD
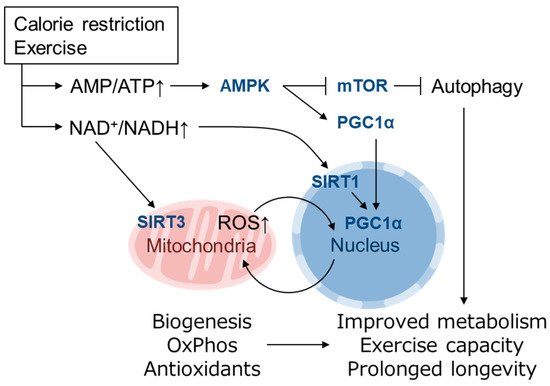
+/NADH and AMP/ATP ratio and a decrease in insulin/IGF signaling or mechanistic target of rapamycin (mTOR) signaling may mediate cellular adaptive responses leading to mitohormesis
[3][39]. The NAD
+-dependent deacetylase Sir2 is reported to promote longevity in yeast
[40][41] as well as
C. elegans and
Drosophila [42][43]. In mammals, there are 7 Sir2 homolog family members, SIRT1-SIRT7, which have diverse functions in various organelles
[39]. Mitochondrial biogenesis induced by a low energy state is mediated by PGC1α, which is a transcriptional cofactor of PPARγ, ERRα, FoxO1 as well as NRF-1
[44][45]. PGC1α is regulated by both transcriptional and posttranslational modifications, the latter of which includes phosphorylation by AMPK (activated by high AMP/ATP ratio) and deacetylation by SIRT1 (activated by high NAD
+/NADH ratio)
[39][44] (). Recently, it was reported that PGC1α activity is also regulated by oxygen availability via Lys224 demethylation by the oxygen-dependent demethylase KDM3A
[46]. The inhibition of mTOR complex 1 (mTORC1) by AMPK as well as amino acid starvation induces autophagy, which is also involved in longevity in yeast, invertebrates and mice
[47]. Mitochondrial biogenesis is also induced by physical exercise
[48][49] and has been proposed to extend lifespan, at least in part, by activating the same pathways
[3]. Increased NAD
+/NADH also activates the mitochondrially localized family member SIRT3, which up-regulates ATP synthesis by activating enzymes in the TCA cycle, ETC and fatty acid oxidation and contributes to antioxidant defense by SOD2 induction via FoxO1 deacetylation
[50] (). Mitochondrial unfolded protein response (UPR
MT) is also thought as a mitohormetic machinery
[51]. In UPR
MT, expression of mitochondrial chaperones and proteases is up-regulated in response to mtDNA depletion
[52] and inhibition of mitochondrial chaperone TRAP1 or protease LONP1
[53]. UPR
MT in nematode is mediated by ATFS-1 as described above
[22], whereas mammalian UPR
MT is thought to be regulated by ATF4, ATF5 and CHOP
[54].
In
C. elegans, skinhead-1 (SKN-1) is the functional homolog of mammalian Nrf1 and Nrf2; however, SKN-1 activation by oxidative stress is mediated by p38 MAPK because Keap1 is absent in
C. elegans [55]. SKN-1 promotes longevity in wild-type worms and is necessary for longevity extension by multiple pathways, including reduced insulin/IGF-1 signaling (DAF2), mTOR inhibition, and dietary restriction
[55]. In contrast, few reports have investigated the role of Nrf2 in the mitohormesis-like response in mammals. Pulliam et al. reported Nrf2 activation in mice lacking surfeit locus protein 1 (
SURF1), an assembly factor of cytochrome c oxidase (COX/Complex IV) encoded in the nuclear genome
[56]. Loss-of-function mutations in the
SURF1 gene lead to mitochondrial disease and Leigh syndrome in humans
[57].
Surf1-KO mice exhibit decreased COX activity in most tissues and reduced endurance capacity but no neurodegeneration symptoms. Interestingly,
Surf1-KO mice are resistant to kainic acid-induced neurotoxicity and show a prolonged lifespan
[56][58].
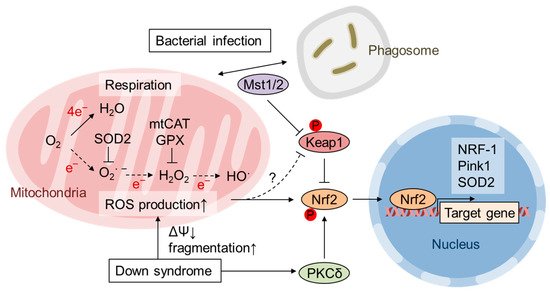
Surf1-KO mice show a decrease in mitochondrial ROS and an increase in mitochondrial number and PGC1α expression in both heart and skeletal muscle.
Nrf2 and
HO-1 expression is elevated in the heart, whereas the expression of UPR
MT genes,
Hsp60,
ClpP, and
Lonp1, is increased in skeletal muscle
[56]. Cox et al. revealed that the temporary depletion of SOD2 expression in embryogenesis potentiates Nrf2 signaling in later developmental stages
[13]. The authors showed that inducible shRNA-mediated SOD2 knockdown during embryonic days 8.5-12.5 increased oxidative stress hallmarks and reduced aconitase activity without newborn lethality. The adapted mouse livers had restored SOD2 expression and aconitase activity and exhibited decreased mitochondrial respiration, decreased ROS levels, and increased mitochondrial content at 4 weeks old. The gene expression profile identified a gene subset with increased expression in which cis elements of PPARγ, PGC1α and Nrf2 were significantly enriched. These responses were also observed in SOD2-knockdown MEFs. When the down-regulated SOD2 expression was restored after transient SOD2-knockdown, MEFs exhibited increased mitochondrial content, Nrf2 target gene expression induction, and reduced maximal respiration and were resistant to menadione-induced oxidative stress. This adaptive response possibly remodels mitochondrial function so that ROS production is reduced by the increased number of mitochondria and restricted respiration rate as well as increased antioxidant defense response
[13]. The molecular mechanisms by which Nrf2 activation is achieved and is maintained for a prolonged period after transient stress stimulation remain to be clarified.
 +1 point
+1 point