1000/1000
Hot
Most Recent
 +1 point
+1 point
Cancer cells typically have elevated proteotoxic stress as a result of genomic instability. The disruption of protein homeostasis causes endoplasmic reticulum stress. If not promptly managed, it could lead to a global decline in cellular function and eventual cell death[1]. This summary of review explores different protein quality control pathways and the translation of drugs targeting proteotoxic stress in haematologic cancers (using multiple myeloma as an example) versus solid cancers (using Triple Negative Breast Cancer as an example).
Cancer cells typically have elevated proteotoxic stress as a result of genomic instability. This property has led to the discovery and development of new anticancer therapies. The disruption of protein homeostasis causes endoplasmic reticulum (ER) stress. If not promptly managed, it could lead to a global decline in cellular function and eventual cell death[1].
The protein synthesis process is extremely prone to errors. About 30% of newly synthesised proteins are degraded by the proteasome within minutes of protein translation in mammalian cells[2]. These rapidly degraded proteins are called defective ribosomal proteins (DRiPs) or rapidly degraded polypeptides (RDPs) [3]. In comparison to normal cells, cancer cells generally synthesise proteins (and therefore DRiPs or RDPs) more rapidly due to increased cell division coupled to cell growth [4]. If DRiPs or RDPs are not removed, the basal proteasome load and cellular stress can increase considerably.
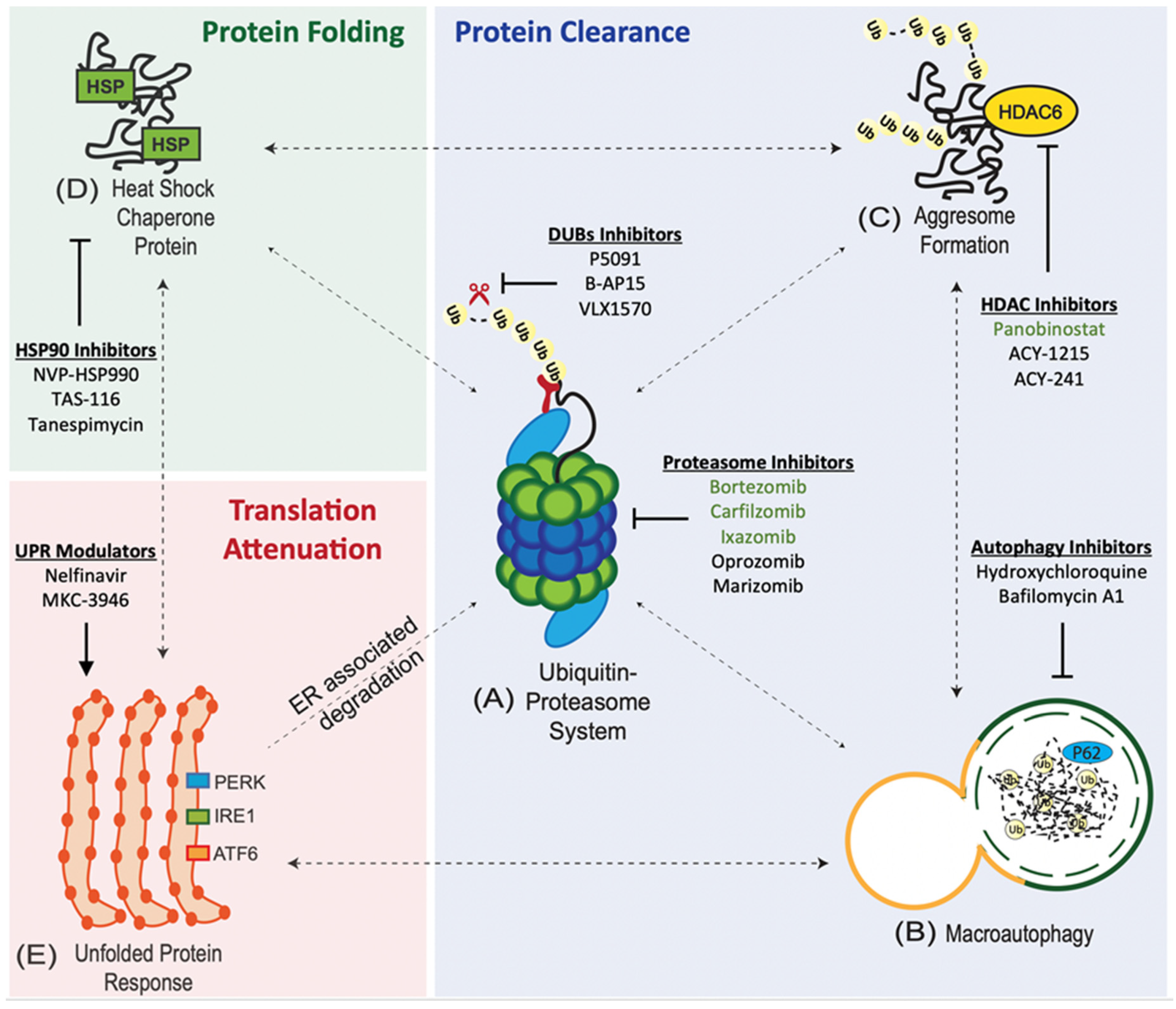
Protein quality control pathways play an important role in tumours due to the intrinsic genetic instability and hostile tumour microenvironment [5]. Cancer cells maintain the integrity of the proteome through an interconnected network of proteostasis pathways.
1. Ubiquitin Proteasome System (UPS). UPS is at the core of proteostasis pathways, and is involved in the degradation of both normal and misfolded proteins [5]. The UPS involves a 3-enzyme-cascade: E1 (activating), E2 (conjugating), and E3 (ligating) enzymes [6]. The targeted (misfolded) proteins are polyubiquitinated or tagged by these 3 enzymes and subsequently degraded by the 26S proteasome [7].
2. Aggresome Pathway. Polyubiquitinated (misfolded) proteins that coalesce to form aggresomal particles are transported towards the microtubule organising center (MTOC) and undergo degradation by autophagy or refolding by the heat-shock protein (HSP) chaperone system [8].
3. Macroautophagy (Autophagy-Lysosome System). Misfolded proteins are taken up by autophagosomes and degraded by lysosomal hydrolases upon fusion with lysosomes [9].
4. Heat-Shock Protein (HSP) Chaperone System. Misfolded proteins can refold with the assistance of HSP chaperones [8].
5. Integrated Stress Response (ISR). Four serine/threonine protein kinases: (i) GCN2 (nutrient deprivation), (ii) PKR (viral infection), (iii) HRI (heme deprivation), and (iv) PERK (ER stress) are activated by cellular stress. The kinases converge on the phosphorylation of eIF2α [10][11].
6. Unfolded Protein Response (UPR). Misfolded proteins are retained in the ER and delivered for proteasome degradation after retro-translocation into the cytosol [12].
The accumulation of misfolded proteins causes ER stress and activates the UPR via three transducers: (i) activating transcription factor 6 (ATF6), (ii) inositol-requiring enzyme 1 (IRE1), and (iii) protein kinase RNA-like ER kinase (PERK) [12].
1. Proteasome Inhibitors (PIs). PIs are the mainstay of MM therapy. Proteasome inhibition exploits the unique biology of MM as myeloma cells have large amounts of misfolded proteins, and hence, high levels of proteotoxic stress due to extensive immunoglobulin synthesis [13][14]. There are 3 classes of PIs: (i) boronates, (ii) epoxyketones, and (iii) γ-lactam-β-lactones (salinosporamide).
2. Autophagy Inhibitors. Dual inhibition of autophagy and UPS, with hydroxychloroquine (HCQ) and bortezomib, has recently emerged as a way to overcome therapeutic resistance to proteasome inhibition [15].
3. Aggresomal Pathway Inhibitors. Panobinostat is a broad-spectrum histone deacetylase (HDAC) inhibitors that has been approved for use in refractory/relapsed MM in combination with bortezomib and dexamethasone [16].
4. Heat Shock Protein 90 (HSP90) Inhibitors. HSP90 inhibitors in preclinical development include NVP-HSP990 and TAS-116 [17][18]. Phase I/II trial of tanespimycin (HSP90 inhibitor) in combination with bortezomib has shown a 27% objective response rate (ORR) [19]. However, further development of tanespimycin was halted due to patent expiry [20].
5. Unfolded Protein Response (UPR) Modulators. UPR is activated by drugs (ER stressors) such as tunicamycin, thapsigargin, and brefeldin A. However, these drugs have limited clinical translation due to their anticipated toxicities based on in-vivo studies [21][22][23]. It has been demonstrated that Nelfinavir, a HIV protease inhibitor, potentiates the anti-MM activity of bortezomib via the activation of the UPR and CHOP-dependent apoptosis [24].
Proteasome Inhibition in TNBC. The effect of proteasome inhibitors against solid tumours like triple negative breast cancer (TNBC) has been quite discouraging. A recent study has demonstrated that inhibition of both the β5 and β2 sites of the proteasome sensitised TNBCcell lines to bortezomib [25]. Paclitaxel, a chemotherapeutic drug, has been shown to regulate the genes involved in the ubiquitin proteasome system in breast cancer. Paclitaxel treatment resulted in an upregulation of proteasome genes and lead to an accumulation of proteasome subunits alpha 4 and beta 1, 26S ATPases 2 and 6 and 26S non-ATPase 14 [26].





