1000/1000
Hot
Most Recent
 +1 point
+1 point
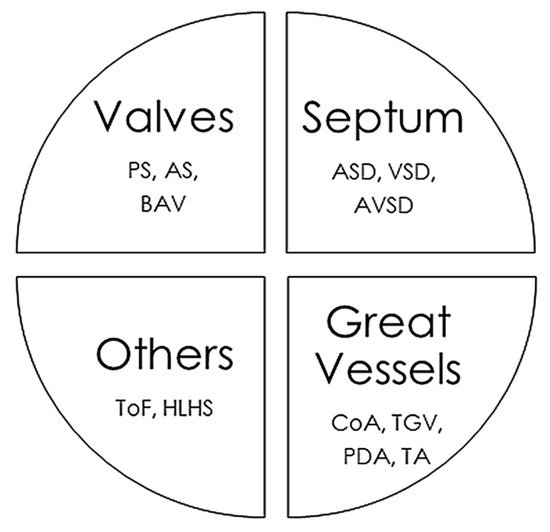
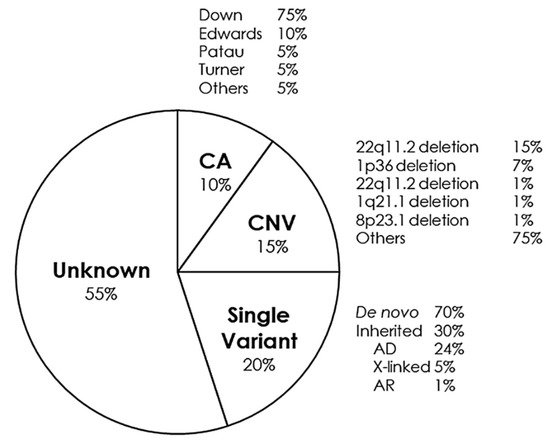
Congenital heart disease is a group of pathologies characterized by structural malformations of the heart or great vessels. These alterations occur during the embryonic period and are the most frequently observed severe congenital malformations, the main cause of neonatal mortality due to malformation, and the second most frequent congenital malformations overall after malformations of the central nervous system. The severity of different types of congenital heart disease varies depending on the combination of associated anatomical defects. The causes of these malformations are usually considered multifactorial, but genetic variants play a key role. Currently, use of high-throughput genetic technologies allows identification of pathogenic aneuploidies, deletions/duplications of large segments, as well as rare single nucleotide variants. The high incidence of congenital heart disease as well as the associated complications makes it necessary to establish a diagnosis as early as possible to adopt the most appropriate measures in a personalized approach. In this review, we provide an exhaustive update of the genetic bases of the most frequent congenital heart diseases as well as other syndromes associated with congenital heart defects, and how genetic data can be translated to clinical practice in a personalized approach.