Hemopexin (Hpx) is a unique internal scavenging glycoprotein that is responsible for the homeostatic maintenance of blood through the regulatory binding of free heme, which eliminates heme’s harmful pro-oxidant and pro-inflammatory potential. Sickle cell disease (SCD) is caused by mutations in the Hb subunit β (HBB) gene coding for the β-globin subunit of the Hb molecule. Specifically, in the series of hemoglobinopathies included under the umbrella term SCD, at least one of the two β-globin subunits of Hb is replaced with the abnormal β-globin hemoglobin S (HbS).
1. Overview
Circulating hemopexin is the primary protein responsible for the clearance of heme; therefore, it is a systemic combatant against deleterious inflammation and oxidative stress induced by the presence of free heme. This role of hemopexin is critical in hemolytic pathophysiology. In this review, we outline the current research regarding how the dynamic activity of hemopexin is implicated in sickle cell disease, which is characterized by a pathological aggregation of red blood cells and excessive hemolysis. This pathophysiology leads to symptoms such as acute kidney injury, vaso-occlusion, ischemic stroke, pain crises, and pulmonary hypertension exacerbated by the presence of free heme and hemoglobin. This review includes in vivo studies in mouse, rat, and guinea pig models of sickle cell disease, as well as studies in human samples. In summary, the current research indicates that hemopexin is likely protective against these symptoms and that rectifying depleted hemopexin in patients with sickle cell disease could improve or prevent the symptoms. The data compiled in this review suggest that further preclinical and clinical research should be conducted to uncover pathways of hemopexin in pathological states to evaluate its potential clinical function as both a biomarker and therapy for sickle cell disease and related hemoglobinopathies.
2. Heme Clearance
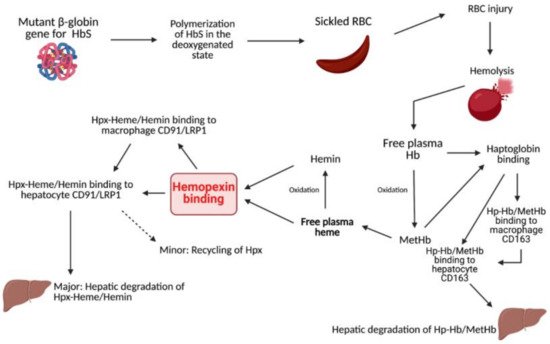
Hemopexin (Hpx) is a unique internal scavenging glycoprotein that is responsible for the homeostatic maintenance of blood through the regulatory binding of free heme, which eliminates heme’s harmful pro-oxidant and pro-inflammatory potential
[1]. Free hemoglobin (Hb) in circulation is oxidized to methemoglobin (metHb), which rapidly degrades to free heme. After metHb degradation, free heme is ultimately bound by Hpx. This binding can occur before or after oxidation to hemin and is predominantly recognized as a 1:1 binding ratio. In the past few decades, a few studies have unveiled evidence of multiple heme binding sites on Hpx, indicating a need for a further evaluation of this association ratio
[2][3][4]. After Hpx–heme complexes are formed, the elimination of heme is mediated by macrophages, typically via the membrane-bound receptor CD91, also known as LRP1. Via signal transduction, CD91 causes the complex relocation of heme to parenchymal cells in the liver for heme catabolism and Hpx recycling. CD91 is also found on the cell membranes of hepatocytes that serve to recognize Hpx-heme complexes and facilitate the subsequent endocytosis of heme into the liver for further breakdown
[5][6]. Hpx exhibits the highest protein-binding affinity in the body for free heme and, consequently, plays a significant role in mediating outcomes after injury or disease (Figure 1). For example, Hpx scavenging of free heme contributes to the protection of numerous cells and regulatory processes, such as macrophages, endothelial cells, and hepatic parenchymal cells, through the alleviation of heme toxicity and the oxidation of lipoproteins
[5]. Additionally, Hpx works with haptoglobin (Hp) to simultaneously combat downstream pro-oxidant and pro-inflammatory damage mediated by the presence of free heme and its predecessor, hemoglobin (Hb). Via receptor-mediated signal transduction and direct Fenton oxidation, heme and Hb, as well as their oxidized respective counterparts hemin and metHb, induce cell damage and hemodynamic conditions that are ultimately deleterious to the prognosis of several pathologies
[7]. By using Hpx and Hp to clear these compounds from circulation, it is possible to reduce the production of reactive oxygen species (ROS) and downstream oxidative and inflammatory damage. It is well documented that heme is a potent inducer of heme oxygenase-1 (HO1) involving the Nrf2 pathway, and the activation of such pathway leads to the increased expression of various other proteins that can mitigate heme-related toxicity. This protection is critical in pathologies that induce increased hemolytic conditions, such as sickle cell disease (SCD).
Figure 1. Summary of heme- and Hb-clearing pathways.
3. Sickle Cell Disease
SCD is caused by mutations in the Hb subunit β (HBB) gene coding for the β-globin subunit of the Hb molecule. Specifically, in the series of hemoglobinopathies included under the umbrella term SCD, at least one of the two β-globin subunits of Hb is replaced with the abnormal β-globin hemoglobin S (HbS). In effect, the mutated β-globin gene leads to polymerized Hb chains in the deoxygenated state. Other mutations of the HBB gene include hemoglobin C, hemoglobin E, or variants leading to underexpressed β-globin subunits. These subsets of the disease with an underexpression of β-globin are referred to as β-thalassemia, which is often comorbid with other hemoglobinopathies. While these pathologies are often seen together, a discussion specifically on Hpx in the case of β-thalassemia was omitted to focus on the hemolytic disorders of SCD. Sickle cell anemia (SCA) specifically refers to homozygosity for the HbS gene, in which patients with SCD produces two abnormal β-globin subunits. In the United States alone, approximately 100,000 Americans are affected, and it occurs in 1 in 1000 to 1400 Hispanic Americans and 1 in 500 African Americans
[8][9][10]. Additionally, SCD is common internationally, with more than 300,000 children born with SCD annually; therefore, it is of particular interest for therapeutic development
[11].
4. Sickle Cell Disease and Hemolysis
The dynamics of Hpx are crucial to outcomes for patients with SCD. Intracellular Hb polymers induce the sickled shape of erythrocytes, which shortens the lifespan of these cells and leads to occlusion of the vasculature, as well as erythrocyte injury
[6]. After erythrocyte injury, there is an excessive degree of extravascular and intravascular hemolysis. The latter leads to an elevated level of free decompartmentalized Hb and heme. Specifically, in SCD, heme exhibits an increased dissociation constant from sickled Hb, further contributing to high levels of free heme
[12]. These processes typically lead to symptoms of pain from vaso-occlusion, including acute lung injury, acute chest injury, pulmonary hypertension, vaso-occlusive crises, and ischemia-reperfusion injury
[13]. The accumulation of free heme and Hb, as well as hemin and metHb, initiate numerous harmful pro-oxidant and inflammatory cascades. These inflammatory cascades include toll-like receptor (TLR) 2 and TLR4, which initiate the broad innate immune response due to their recognition of damage-associated molecular patterns (DAMPs)
[14][15][16]. These response cascades ultimately lead to a decreased quality of life for patients with SCD because they cause increased severe acute lung injury, vaso-occlusion, and pain crises, among other conditions. It has been reported that hyperalgesia, one of the most prominent chronic manifestations of SCD, is directly exacerbated by TLR4 activation via neuroinflammation, microglial activation, and endoplasmic reticulum stress
[13]. This evidence has been corroborated by analyses of patients with SCD showing increased amounts of free heme in circulation
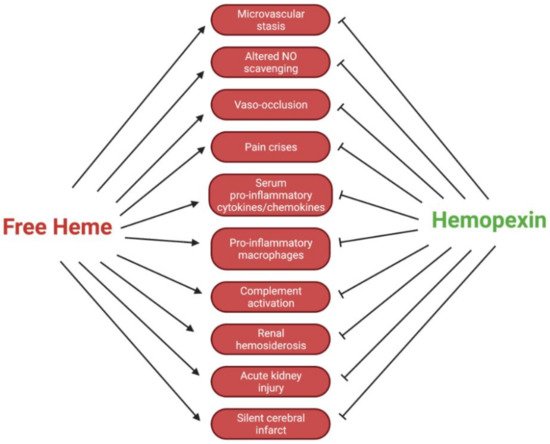
[17]. In reference to transfusion therapies in SCD patients, reducing the activation of the innate immune response against free heme by using Hpx could hypothetically mitigate the dependence on immunosuppressive therapies. Clinically, this approach is dynamic and must be further studied before application. Additionally, the altered scavenging of vasodilating nitric oxide (NO) by free Hb could lead to worsened vasoconstriction. With narrowed systemic vasculature and pathologically sticky erythrocytes, patients are prone to painful and deleterious vaso-occlusion (Figure 2).
Figure 2. Regulatory effects of Hpx in SCD pathologies.
In addition to the pain and pulmonary symptoms associated with sickled cells resulting from TLR4 activation, patients with SCD experience an increased incidence of ischemic events. Most likely due to the compact vasculature of the brain in combination with the increased stickiness of sickled erythrocytes, ischemic pathologies are predominantly seen in children and the elderly
[10][18][19]. It is well-documented that Hpx plays a neuroprotective role after ischemic injury; therefore, it should be evaluated as a dynamic therapeutic target, specifically in patients with SCD
[20][21][22][23]. According to Li et al., the local expression of Hpx by neurons contributes to protection from free heme through the induction of HO isoenzymes, namely HO1
[20]. The upregulation of HO1 in astrocytes and fibroblasts in the ischemic penumbra exhibit protective effects against ischemic insults by reducing oxidative stress and apoptosis. In Hpx
−/− mice, there is evidence of increased oxidative damage, infarct volume, and overall neurological deficiency after ischemic stroke
[20][23]. This finding indicates that Hpx plays a multifaceted role in neuroprotection against ischemic events commonly associated with SCD. Here, we summarize the current understanding of Hpx dynamics in sickle cell-associated disorders, specifically those worsened by the presence of free heme.
5. Conclusions
There is ample evidence that Hpx is a unique, multitasking protein that plays a significant role in heme-mediated protection against microvascular stasis, vaso-occlusion, and lipid oxidation, specifically in patients with SCD. The findings from this review support the argument for continued preclinical studies and possibly subsequent clinical studies regarding the use of Hpx as an intervention for SCD pathologies. Although much remains to be elucidated before suggesting a formal Hpx therapeutic regimen, the deleterious effects of free heme and the potentially cytoprotective effects of Hpx have become apparent in recent research in this field.
 +1 point
+1 point