DNA repeat expansion disorders are a group of neuromuscular and neurodegenerative diseases that arise from the inheritance of long tracts of nucleotide repetitions, located in the regulatory region, introns, or inside the coding sequence of a gene. Although loss of protein expression and/or the gain of function of its transcribed mRNA or translated product represent the major pathogenic effect of these pathologies, mitochondrial dysfunction and imbalance in redox homeostasis are reported as common features in these disorders, deeply affecting their severity and progression.
1. Introduction
Microsatellites are stretches of DNA abundantly interspersed in the genome of prokaryotes and eukaryotes
[1][2], including humans, where they account for the 3% of the genome
[3]. Their structure consists of short, tandemly repeated duplications of 1–6 base pairs, spanning between 20–100 bases
[4] and primarily consisting of mono- and dinucleotides, although tri-, tetra-, penta-, and hexa-nucleotides microsatellite classes are present
[4][5]. Their location is ubiquitous, occurring both in protein-coding as well as in non-coding DNA regions, with preference for the latter
[6], and their functions range among several biological regulatory processes
[7], including alternative splicing
[8], transcription start/end site selection
[9][10], nucleosome packaging
[11], and methylation
[12]. One of the most peculiar characteristics of microsatellites is their tendency to mutate. While 10
−9 is the rate of mutations occurring in non-repetitive region of the DNA, microsatellite mutation rate ranges between 10
−2 and 10
−6 [13][14]. Because of this, microsatellites are highly polymorphic, as the number of the repeats in a given locus is relatively unstable and frequently varies between individuals
[15]. Due to this feature, deregulated microsatellite expansions are widely known to be the triggering cause of many neurological and neuromuscular diseases and, to date, more than 30 disorders are known to be caused by aberrant expansion of repetitive DNA sequences
[16]. Although trinucleotides repeats, in particular GAA, CGG, CAG, and CTG, are commonly known to be responsible for the pathologic manifestation of nucleotide expansion disorders, also tetra- and penta-nucleotides expansions are disease causing
[17], thus these diseases are collectively classified as disorders of DNA unstable repeat expansions
[18]. Some peculiarities join this variegated group of pathologies: (i) The disease-causing expansion repeats are more unstable in the affected population, with a higher tendency to expand or to contract with respect to the polymorphic repeats of normal people, even if contraction events are rarer
[16]. This also occurs as the repeats reach the threshold limit between a normal condition and the pathological state. Thus, even in unaffected families, de novo mutations can determine clinical manifestations
[19]. (ii) The dynamic changes in the length of expansions are so marked that differences among patients, as well as in different tissues of the same affected proband, are common
[16]. (iii) The more the expansions are transmitted from generation to generation, the earlier the disease symptoms appear in the newly affected individuals, a phenomenon known as clinical anticipation
[20][21][22]. (iv) In most disorders, the length of repeat expansions influences the phenotype severity of affected individuals
[23][24][25][26][27][28][29]. Consequently, different defects for a given disease can be expressed among patients, including onset of symptoms and co-morbidities. For example, the incidence of diabetes in Friedreich’s ataxia (FA) ranges between 8–32% of patients
[30][31][32], but the risk of developing is directly correlated to the number of GAA repeats in the frataxin (
FXN) gene
[33]. In the same way, in myotonic dystrophy (DM), patients carrying small CTG repeats (i.e., between 50–99) are asymptomatic or develop mild defects, such as cataracts, while a severe phenotype occurs in patients with 100–200 repeats
[34].
Beside these common features, a high grade of diversity characterizes DNA expansion disorders, as the DNA expanded tracts can affect genes encoding proteins with different roles. Therefore, three different classes have been distinguished, by assembling disorders on the basis of which defects arise from expansions. The first class groups the diseases that are determined by a protein loss of function and are inherited by autosomal recessive or x-linked manner
[19]. Typical examples are Fragile X (FXS) or FA, where CGG or GAA repeats determine the loss of the expression of the fragile X mental retardation protein (FMRP) and FXN, respectively
[35][36]. The second group belongs to disorders characterized by an autosomal dominant inheritance and in which a protein gain of function occurs
[18]. PolyQ diseases, for instance, are determined by CAG expansions in the coding region of 9 distinct genes that lead to the formation of glutamine residues in the final peptide of their encoded product
[37]. PolyQ tracts exert a toxic effect mainly by causing aberrant nuclear and cytoplasmic protein aggregation and trapping transcription factors
[38][39], chaperons, and proteins belonging to the ubiquitin–proteasome system (UPS)
[40]. The third group of disorders are characterized by gain of function involving the transcribed RNA. DM1 and DM2 are respectively caused by the aberrant insertion of CTG and CCTG expansion repeats in the 3′ untranslated region (UTR) of dystrophin myotonic protein kinase (
DMPK)
[41] and in the first intron of zinc-finger protein 9 (
ZNF9)
[42]. Similarly, the CGG triplet expansion in
FMR1 gene, ranging between 60–200 triplets, causes the fragile X–associated tremor ataxia syndrome (FXTAS)
[43]. The pathogenic activity, both in DM and in FXTAS, lies on of the respective expanded mRNA molecules, which are able to sequestrate RNA binding proteins, such as muscleblind-like (MBNL) proteins in DM1, determining splicing alterations and impairments in protein expression
[44][45][46][47].
Oxidative stress has been widely reported to play a prominent role in neurodegenerative diseases
[48][49], including disorders caused by DNA expansion repeats (). Here, we report the most recent evidences connecting ROS imbalance and DNA expansion disorders, with particular emphasis to the pathway primarily involved in the regulation of cellular antioxidant response, the NF-E2 p45-related factor 2 (NRF2) signaling pathway.
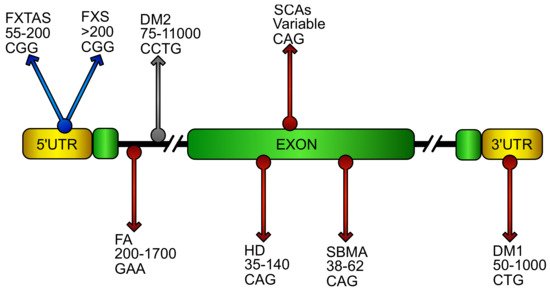
Figure 1. Putative location and sequence of DNA expansions in repeat disorders. Schematic representation of an ideal gene showing DNA repeat expansions that cause diseases. Name of the relative disorder, number of pathogenic repeats, and its sequence are reported in the gene region were the repeats stem in the pathology. The grey arrow represents pathology where oxidative stress has been poorly investigated. Blue arrows characterize diseases with oxidative stress contributions. Red arrows identify pathologies in which NF-E2 p45-related factor 2 (NRF2) involvement has been reported. (FXTAS, fragile X–associated tremor ataxia syndrome; FXS, fragile X syndrome; FA, Friedreich’s ataxia; DM1/DM2 myotonic dystrophy; HD, Huntington’s disease; SCAs, spinocerebellar ataxias; SBMA, spinobulbar muscular atrophy).
2. NRF2 Pathway and Its Regulation
NRF2 is a transcription factor belonging to the cap ‘n’ collar (CNC) basic leucine zipper (bZip) proteins
[50][51]. In the nucleus, it dimerizes with the small musculoaponeurotic fibrosarcoma (sMAF) proteins, particularly with F, G, and K isoforms
[52][53]. The NRF2-sMAF heterodimer binds to specific 16 base long DNA stretches (the antioxidant responsive elements, ARE), acting as enhancer for gene transcription
[54]. NRF2 is able to regulate the expression of at least 250 genes
[55] and, besides being the master regulator of cellular antioxidant defense, its activity participates to the modulation of different cellular processes, including metabolism, survival, differentiation, inflammation, mitochondrial biogenesis, and mitophagy
[56][55][57][58][59][60][61][62]. For this reason, the NRF2 activity and expression are subjected to a tight and fine-tuned control mechanism, to avoid unwanted gene expression upregulation and, at the same time, determining a fast response in case of need. Upon oxidative stress, the induction of NRF2 occurs by regulating its stability and localization in the cell
[63], and modulating the amount of its mRNA transcript
[59][64].
Under physiological condition, NRF2 has a short half-life, spanning between 15–40 min, and its cellular localization is restricted to the cytoplasm
[65]. Soon after translation, NRF2 interacts with the ubiquitin ligase adaptor KEAP1 (Kelch-like ECH-associated protein 1) that sequesters the transcription factor and mediates its proteasomal degradation
[66]. In parallel, free NRF2 can be phosphorylated by the GSK3β kinase, which increases the NRF2 proteasomal-mediated turnover
[67][65]. These two mechanisms work in conjunction to regulate NRF2 activity, in response to different cellular cues. Under redox imbalance, the KEAP1–NRF2 interaction is disrupted, as result of ROS-induced conformational changes of KEAP1
[68]. Conversely, the activation of growth factor receptors determines the AKT/PI3K-induced inhibitory phosphorylation of GSK3β, thus allowing NRF2 accumulation
[69]. This regulatory system points to the NRF2/ARE axis as one of the most important signaling pathway in cells. Indeed, being ARE sequences implicated in the regulation of more than 1% of human genes
[65], impairments of NRF2 signaling network may interfere with multiple cellular processes and determine redox imbalance, a condition commonly encountered in cancers
[70][71][72] and in neurological disorders. Re-establishing NRF2 signaling homeostasis can be essential to improve the pathological phenotypes, especially in neurodegenerative diseases
[73][74][75].
Recent evidences increasingly highlight a dual role of NRF2 in the diseases’ pathogenesis. In cancer, for instance, the activation of Nrf2 appears correlated with progression and chemo-resistance, and its downregulation has attracted growing attention as alternative cancer therapy
[76]. Several studies have clearly demonstrated that the hyper-activation of the NRF2 pathway may create an environment favoring the survival of malignant cells, protecting them against oxidative stress, chemotherapeutic agents, and radiotherapy
[77][78]. Indeed, although a transient NRF2 activation in response to stress may be beneficial for health, a persistent induction can confer therapeutic resistance in cancer cells and more aggressive tumorigenicity, leading to poor prognoses in patients. In this light, the inhibition of NRF2 is a promising therapeutic approach in cancer and NRF2 inhibitors are being actively developed
[79]. Contrariwise, NRF2 appears inhibited in many neurodegenerative disorders, such as Huntington’s disease, Alzheimer’s disease, amyotrophic lateral sclerosis, multiple sclerosis and FA, where its activation has been proven mitigating pathogenic processes by upregulating antioxidant defenses, decreasing inflammation, and improving mitochondrial function
[58][80][81][82][83]. Therefore, a dual face of Nrf2 in cancer and neurodegenerative diseases has to be recognized, making its role in the pathogenesis’ mechanisms even more attractive.
3. Examples of Oxidative Stress in Loss of Function DNA Expansion Disease
3.1. Friedreich’s Ataxia (FA)
(FA) is an autosomal recessive neurodegenerative disease caused by a homozygous GAA trinucleotide repeat expansion in the first intron of the FXN gene, encoding for the mitochondrial FXN protein
[36][84]. The GAA repeated expansions cause histones deacetylation and abnormal DNA conformation, leading to decreased mRNA levels and FXN amount
[85]. FA is clinically characterized by progressive ataxia, diabetes, cardiomyopathy, skeletal deformations, altered central and peripheral nervous system with lesions in the dorsal root ganglia, dentate nuclei of the cerebellum, corticospinal tracts, and sensory peripheral nerves
[86][87][88]. Actually, the FXN function is still unclear, although it is well known to be involved in iron–sulphur cluster biogenesis and in heme biosynthesis. The FXN deficiency increases the mitochondrial iron content, altering activities of iron–sulphur (Fe-S) cluster enzymes in mitochondria and causing oxidative stress in affected tissues
[89][90][91]. Oxidative stress is a leading hypothesis in the pathogenesis of FA, since the identification of the gene in 1996 and later supported and confirmed by several studies in human and mouse FA models
[92][93][94][95].
Several studies have demonstrated an impairment of the NRF2 pathway in FA
[96][97][98][99] and alterations of systemic redox markers have been evidenced in patients. An increased oxidative damage on nuclear and mitochondrial DNA has been found in peripheral blood cells, together with high levels of plasma malondialdehyde and of urine 8-hydroxy-2-desoxiguanosine
[100][101]. A decrease of glutathione levels and of antioxidant enzymes activities (SOD and GST) have been also reported in fibroblasts and in blood of patients
[102][103]. Recently, lipid peroxidation and ferroptosis have also been suggested as responsible for the FA pathophysiology
[104][105][106]. It is important to note that two ferroptosis-triggering enzymes (GPX4 and cysteine/glutamate transporter system, xC-/xCT) are downstream targets of NRF2
[107]. For all these findings, NRF2 has recently attracted attention for novel therapeutic strategies in FA
[63][74][108][109]. Among the main pharmacological NRF2 activators, we can mention the Sulforaphane (SFN), a natural blood–brain-barrier permeable antioxidant, and the dimethyl fumarate (DMF), an ester of fumaric acid recently approved for the treatment of multiple sclerosis
[110][111][112][113] and promising for adrenoleukodystrophy
[114]. The efficacy of these NRF2 inducers has been verified on several FXN deficient models
[59][64][115], where they significantly increased NRF2 mRNA levels
[64], re-balanced the GSH/GSSG ratio
[64][115], and up-regulated the FXN gene expression
[64][115][116][117] ().
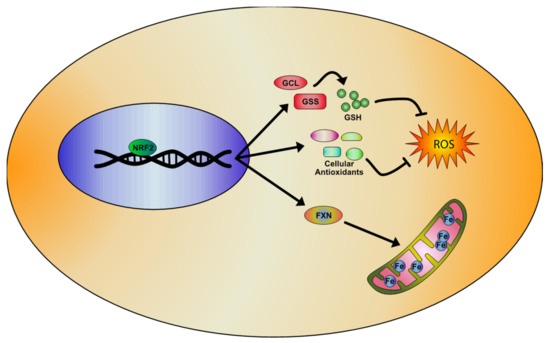
Figure 2. Representative model of the NRF2 signaling pathway activation in Friedreich’s Ataxia (FA), based on literature evidences. NRF2 inducers determine the activation of antioxidant genes transcription and the upregulation of enzymes involved in the regulation of glutathione (GSH) expression, rebalancing the unpaired GSH/GSSG ratio and reducing oxidative stress and lipid peroxidation. Importantly, NRF2 also increases frataxin (FXN) levels, thus partially rescuing the mitochondrial defects observed in FA pathology.
Currently, clinical FA trials are focused on improving mitochondrial function and reducing oxidative stress
[118]. Idebenone (Raxone
®/Catena
®), for instance, is proven to be effective on the mitochondrial function. But, despite an initial optimism on its cardiac impact, the neurological benefit in FA is still under evaluation
[119][120][121]. RTA408 (Omaveloxolone), a specific NRF2 inducer
[122], is currently being tested in a 12 months placebo-controlled trial (
www.clinicaltrials.gov). EPI-743 (vatiquinone), another highly promising drug for FA
[123], was approved in 2011 for children with genetically confirmed inherited respiratory chain diseases, but still not clinically tested in FA, although it has been proven to activate NRF2 and increase the expression of FXN in fibroblasts of FA patients
[64].
4. Examples of Oxidative Stress in CAG/polyQ diseases
4.1. Huntington’s Disease (HD)
Huntington’s disease (HD) is a progressive, autosomal dominant neurodegenerative disease with defects in the striatum, cerebral cortex, and thalamus [124][125]. The HD disorder is caused by the abnormal expansion of the nucleotide triplet CAG in the gene coding for the protein huntingtin [126]. In the huntingtin gene (HTT) of healthy subjects, the number of trinucleotides CAG repeats varies from 1 to 34, while in HD patients, the CAG triplet expansion ranges between 35–140 repetitions [127]. Clinical features of HD include progressive motor dysfunction, psychiatric disturbance, cognitive decline, dystonia, bradykinesia, and dementia, ultimately leading to death within approximately 15–20 years from the age of onset [128]. The genetic abnormality in the HD gene leads to the formation of a mutant huntingtin protein (mHtt), which is normally involved in the vesicle transport and represents a scaffold for the autophagic machinery [129][130]. The mutant protein exhibits toxic properties, leading to protein aggregation, transcriptional dysregulation, defective energy metabolism, chronic inflammation, and oxidative stress [131][132][133][134]. Inflammation, mitochondrial dysfunction, and oxidative stress are some of the key pathways persistently abnormal in mouse models of HD and in autoptic tissues of patients.
Several pharmacological HD mice models have been developed, resembling defective neuro-motor functions described in human HD patients [135][136][137][138] and supporting oxidative damage as a pathogenic mechanism underlying neurodegeneration in this disease [139][140]. Increased markers of oxidative stress, mitochondrial failure, and chronic inflammation have been found in brain tissue of HD patients. High levels of malondialdehyde, 8-hydroxy-deoxyguanosina, and carbonyls, and lower levels of GSH, SOD1, and GPX, have been detected in plasma and red blood cells of patients [141][142]. Additionally, mitochondrial DNA damage, low levels of oxidative phosphorylation enzymes, and iron-mediated mitochondrial impairment have been shown in autoptic brain tissues of patients [139][143]. In addition, increased amounts of circulating pro-inflammatory cytokines have been reported in patients, whose levels correlated to the severity of the disease [142]. Numerous studies have been focused to reduce oxidative damage in HD by using antioxidants (alpha-tocopherol, CoQ10, vitamin E, vitamin C, N-acetylcysteine (NAC), lipoic acid [144][145][146][147][148][149][150]). Nevertheless, these compounds have shown a moderate effectiveness in counteracting oxidative stress in mouse models, thus leading to hypothesize that a pharmacological upstream activation of NRF2 should be required. Recently, the potent NRF2 inducer SFN has been tested, showing increased mHtt degradation and a significant reduction of cytotoxicity by the NRF2-mediated activation of the ubiquitin–proteasome system [151]. The SFN pre-treatement ameliorated behavioral impairments and reduced pro-inflammatory cytokines in the striatum of a 3-nitropropionic acid (3-NP) mouse model by attenuating neuroinflammation and oxidative stress [151]. High susceptibility to oxidative stress has been also found in human HD neural stem cells, where the genetic correction of the disease-causing mutation restored the redox balance [152]. The protective effect of NRF2 activation in HD patients has been further confirmed in primary monocytes, where the NRF2 induction inhibited the release of pro-inflammatory cytokines (IL-6, IL-1, IL-8, and TNFα) [152]. As in other neurodegenerative diseases, also in HD has been hypothesized a role for ferroptosis in the pathogenic mechanism, mainly as a consequence of increased iron levels that were detected in brain regions of patients [143][153]. Therefore, even in HD, NRF2 can represent a strategic therapeutic target, for its ability in preventing iron overload and regulating ferroptosis-related genes expression [154]. DMF, for instance, exerted beneficial effects on survival and motor functions in R6/2 and YAC128 models of HD, preserving the neuronal integrity in striatum and motor cortex, and slowing degeneration [155]. Additionally, gintonin (GT), a ginseng-derived lysophosphatidic acid receptor ligand, was effective on the NRF2 pathway in the striatum of 3-NPA mice, by protecting the mitochondrial function and reducing the expression of inflammatory mediators (cytokines, COX-2, and iNOS) [156].
5. Examples of Oxidative Stress in RNA Gain of Function Expansion Disease
5.1 Myotonic Dystrophy (DM)
DM is an autosomal dominant disorder, which arises from 2 different mutations: DM1, determined by 50-1000 CUG triplets in the 3′UTR of DMPK gene and DM2, caused by 75– 11000 expansions of the tetranucleotide CCTG in the first intron of ZNF9. DM1 and DM2 are multisystemic diseases sharing a common symptomatology characterized by myotonia, muscular dystrophy, cardiac defects, cataracts [157], and neurological manifestations [158][159]. Unlike DM1, DM2 does not show congenital forms [160]. DM shows a marked somatic instability of repeat expansions that, in DM1, are reported to increase of about 50–80 repeats per year and, in DM2, appear to be even more pronounced [157][161][162]. Depending on the repeat length, the severity of DM1 and the onset of the pathology range from “mild” manifestation (baldness and cataracts) to a “classic” or “juvenile” form, with worse symptoms [19]. On the contrary, although the same clinical heterogeneity is observed in DM2, the pathologic onset and disease severity do not seem to depend on the size of expansions in this disorder [163]. Clinical anticipation, prominent in DM1 [164], appears mildly in DM2 [165].
Different hypotheses have been proposed to explain the pathogenic mechanism in DM. Early studies suggested that the pathological defects observed in DM1 could be determined by the decrease of DMPK expression, mediated by CUG expansions [166] and/or by the trans-acting effect of the expanded mRNA, able to reduce the processing of WT DMPK mRNA [167]. Clinical similarities led to support a common pathogenic mechanism for DM1 and DM2 and, to date, an RNA toxic gain of function is the most credited. In particular, both CUG triplet containing DMPK mRNA and spliced ZNF9 intron1 containing long CCTG sequences are able to sequestrate in the nucleus the splicing factors MBNL1 and 2 [168][169] and, at the same time, to raise the RNA binding activity of CUG-binding protein 1 (CUG-BP1 or CELF1) [170]. This changes the cellular alternative splicing output, determining the defects observed in the disease [171].
Many of the clinical features showed in DM, including myotonia, progressive muscle weakness, cataracts, frontal alopecia, and cognitive decline, suggest an increased susceptibility to oxidative stress in this pathology, as observed in premature and accelerated aging [172]. While in DM2, oxidative stress is still poor investigated, a pathogenic involvement of ROS has been evidenced in DM1. Increased sensitivity to oxidative stress and strong activation of the pro-apoptotic p38 and JNKs pathways have been reported in the C2C12 cell line transfected with human mutant MDPK containing a variable number of CTG repeats [173]. On the contrary, in cells having only 5 CTG repeats, ERKs were preferentially activated [174]. Moreover, studies performed in DM patients have demonstrated an increase of lipid peroxidation and ROS levels, with a parallel decrease of the antioxidant CoQ10 content [175]. Increased oxidative stress and ROS-induced inflammation are known to produce cognitive dysfunctions [176][177] and depressive behaviours [178][179], conditions observed in the MBNL2 KO mouse model of DM1 [180]. In these mice, the chronic administration of methylphenidate (MPH) was able to partially rescue the cognitive defects and depressive-like behaviours, and to reduce the reactive microglia and pro-inflammatory cytokine IL-1β levels [180]. The treatment with MPH was shown to increase NRF2 gene expression in the hippocampus of MBNL2 KO mice and the brain-derived neurotrophic factor (BDNF) levels, which regulates NRF2 nuclear translocation by means of an ERK/PI3K-dependent activation [181]. These findings suggest that the rescue of behavioural defects in MBNL2 KO mice may depend on NRF2-mediated reduction of oxidative stress and inflammation. In line with this, it is important to note that in NRF2 KO mice, an increase of the serum level of pro-inflammatory cytokines and a decrease of the BDNF expression have been reported in association to a depressive-like phenotype [182]. In the same way, NRF2 activation is able to reduce depression and serum content of pro-inflammatory markers induced by lipopolysaccharide (LPS) injections in mice [183]. Cognitive defects [158][159] and depression [184], together with serum increased concentration of the pro-inflammatory IL-6 [185] have been found in DM1 patients, thus the pharmacological NRF2 induction could be very promising in this disease.
 +1 point
+1 point