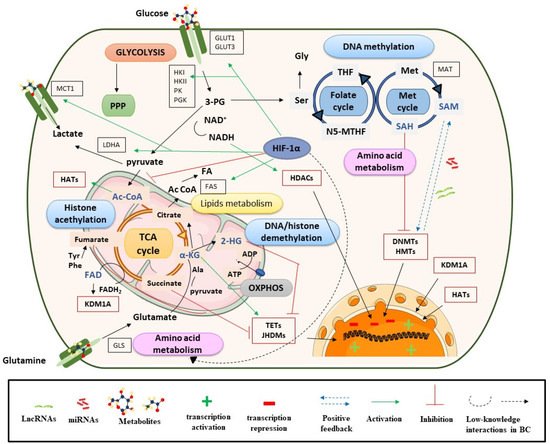
Cancer cells alter their metabolic and nutrient uptake pathways during tumor initiation, growth, and metastasis through a tightly regulated program of metabolic plasticity. This allows them to sustain the energetic and biosynthetic demands of cell proliferation and to adapt to hostile and ever-changing environments
[24]. Epigenetic modifiers act on metabolic gene expression to induce changes in biochemical pathways, and many of the chemical modifications in DNA and histones derive from intermediates of cellular metabolic pathways. This indicates that fluctuations in metabolic concentrations affect the deposition and removal of chromatin modifications. Emphasizing on this last issue, several mechanisms have to be considered, such as: (i) the alteration of specific metabolites’ concentrations that act as epigenetic cofactors or substrates; (ii) the generation of oncometabolites which act as inhibiting or activating epigenetic enzymes; and (iii) the translocation of metabolic enzymes and metabolites into the nucleus
[20]. Below, we address these regulation processes in the context of BC.
2.1. Metabolites and DNA/Histone Methylation Processes
DNA methylation is one of the most studied epigenetic mechanisms in cancer, including BC. Methylation can be produced directly in promoter regions of cancer-related genes (CpG islands) or in residues of histones, and this can control DNA accessibility and regulate gene expression. In DNA or histone methylation processes, the availability of methyl groups is essential for the action of histone methyltransferases (HMTs) and DNA methyltransferases (DNMT)
[20]. In this context, the methionine and folate cycles, as well as the metabolites involved in these pathways (serine, methionine, and the cofactor S-adenosyl-methionine (SAM)), have an important role in supplying one-carbon groups
[20] and they are closely related to DNA methylation processes (). High levels of these metabolites have been found in BC samples and are postulated as candidate biomarkers of BC
[25][26]. SAM provides methyl groups that release S-adenosyl-homocysteine (SAH), an inhibitor of DNMTs and HMTs. Therefore, the SAM/SAH ratio is a major determinant of chromatin methylation. It is known that an increased SAM/SAH ratio correlates with hypermethylation of tumor suppressor genes and inappropriate silencing, whereas a decreased SAM/SAH ratio contributes to reduced methylation at the promoters of oncogenes ()
[27].
Figure 1. Metabolism controls epigenetic enzymes in BC. Ac CoA: acetyl coenzyme A; ADP: adenosine diphosphate; α-KG: alpha-ketoglutarate; ATP: adenosine triphosphate; DNMTs: DNA methyltransferases; FADH2: flavin adenine dinucleotide; GLS: glutaminase; GLUT: glucose transporter; HADAC: histone deacetylase; HATs: histone acetyltransferases; 2-HG: 2-hydroxyglutarate; HIF-1α: hypoxia-inducible factor subunit alpha; HMTs: histone methyltransferases; HK: hexokinase; JHDMs: JmjC-domain-containing histone demethylases; KDMs: lysine demethylases; LDHA: lactate dehydrogenase A; Met: methionine; NADH: nicotinamide adenine dinucleotide; N5-MTHF: 5-methyltetrahydrofolate; OXPHOX: oxidative phosphorylation; 3-PG: 3-phosphoglycerate; PGK: phosphoglycerate kinase; PI3K: phosphoinositide 3-kinases; PK: pyruvate kinase; PPP: pentose phosphate pathway; SAM: S-adenosyl methionine; Ser: serine; TCA cycle: tricarboxylic acid cycle; TETs: ten eleven translocation enzymes; THF: tetrahydrofolate.
Demethylation reactions are also susceptible to metabolic fluctuations of TCA cycle intermediates, such as α-KG, succinate, fumarate, and acetyl-CoA, which have been postulated as BC biomarkers
[26][28]. They act on chromatin-modifying enzymes such as the 2-oxoglutarate-dependent dioxygenases (2-OGDO) family, which include ten eleven translocation (TET) enzymes, and the Jumonji (JHDMs) family of histone demethylases ()
[29][30][31]. These enzymes catalyze the hydroxylation and demethylation of proteins and nucleic acids and play an important role in epigenetic processes. α-KG acts as a positive cofactor of 2-OGDO, thus elevated levels of α-KG from glucose and glutamine catabolism would promote demethylation processes that would influence BC epigenetic landscapes by relaxing chromatin and activating oncogene expression. Additionally, α-KG is a substrate of prolyl hydroxylase (PHDs), a type of protein that regulates hypoxia-inducible factors (HIFs)
[32][33]. HIF subunit alpha (HIF-1α) regulates various processes and, under hypoxic conditions, can promote cancer cell survival. In the presence of oxygen, PHD proteins hydroxylate proline residues on HIF-1α, which leads to HIF-1α ubiquitination by Von Hippel–Lindau tumor suppressor protein (pVHL) and its proteasomal degradation
[34]. In the context of cancer, most tumors have hypoxic regions and, in this case, HIF-1α is stabilized and triggers changes in glycolysis, nutrient uptake, waste handling, angiogenesis, apoptosis, and cell migration, which promote tumor survival and metastasis
[34].
In BC, HIF-1α has a predictive and prognostic role; its overexpression is known to stimulate angiogenesis and can lead to a poor prognosis for patients
[35]. HIF-1α is also closely linked to metabolism and regulates glycolysis, fatty acid, and amino acid pathways. HIF-1α increases glucose uptake by upregulating glucose membrane transporters (GLUT1 and GLUT3), which has been correlated with BC progression and poor overall survival
[36] ().
Furthermore, HIF-1α upregulates lactate dehydrogenase A (LDHA) to promote lactate production, regenerates nicotinamide adenine dinucleotide (NAD+), and increases the transcription of lactate transporters such as monocarboxylate transporter 1 (MCT1)
[37][38]. This, in addition to high levels of hexokinase (HK), promotes a glucose flux towards pyruvate and lactate generation
[39]. Previous studies have shown that lactate levels are related to BC progression and invasive or metastatic BC is known to have higher levels of this metabolite
[38][39]. These metabolic processes would be in concordance with the Warburg effect
[40]. Regarding lipid metabolism, several studies in cancer, including BC, have shown that HIF-1α increases the availability of fatty acids by regulating the action of fatty acid synthase (FAS), increasing fatty acid transport and reducing fatty acid oxidation
[41]. In addition, HIF-1α plays an important role in amino acid metabolism, particularly in glutamine availability ().
Cancer cells use glutamine as an energy substrate, a precursor of fatty acids, a donor of carbon and nitrogen for generating nucleotides or other amino acids, and to maintain the poll of intermediate metabolites such as acetyl-CoA or α-KG. These last metabolites are important for the anaplerotic reactions of the TCA cycle, but also for epigenetic processes by the effect that they have on HAT and HDM
[34]. Due to the role that glutamine has in BC cells, several studies have investigated the inhibitory action that lncRNAs (e.g., lncRNA-p21) or miRNAs (e.g., miR-1, miR-1-3p, miR-9, miR-129) exert on GLS regulation (more details in
Section 3.1.2)
[42][43].
On the other hand, fumarate hydratase (FH) and succinate dehydrogenase (SDH) genes are mutated in many human cancers, including BC, which leads to the accumulation of their substrates, fumarate and succinate, respectively
[30]. This is consistent with metabolomic analyses, which have shown increased levels of these metabolites in BC
[28][44], but also with transcriptomic studies that have shown a strong deregulation of TCA cycle genes
[14]. Among others, succinate, fumarate and α-KG are considered oncometabolites. This term refers to metabolites that are significantly elevated in tumor cells compared with control cells
[45]. Succinate and fumarate, together with 2-hydroxyglutarate (2-HG), can inhibit PHDs activity under normoxic conditions
[31]. Hence, succinate and fumarate could act as competitors of α-KG, inhibiting JHDMs and TET activity, and acting on bladder tumor biology through a profound impact on epigenetic effector activity
[46][47][48] (see ). In conclusion, tumor-gene expression is regulated by epigenetic enzymes, the activity of which is dependent on metabolite availability (substrates).
2.2. Metabolites, Histone Acetylation Processes and Sirtuins
Another important metabolite, acetyl-CoA, is synthetized in several metabolic pathways (mitochondria, cytosol, and nucleus) from several sources, namely pyruvate, acetate, fatty acid β-oxidation, and amino acid catabolism. Metabolomic studies have reported elevated levels of acetyl-CoA in BC
[28], and have particularly highlighted the role of glutamine as a substrate for acetyl-CoA synthesis
[14]. Additionally, upregulated expression of acetyl-CoA synthase enzymes, such as ATP citrate-lyase (ACYL) or acetyl-CoA synthetase short chain family (ACSS), has been frequently found in BC cells, and some studies have reported the importance of ACSS3 for histone acetylation
[49]. Acetyl-CoA acts as a cofactor which modulates kinetic and binding parameters of histone acetyltransferases (HATs). Nevertheless, CoA, the product of histone acetylation reaction, acts as an inhibitor. Therefore, the acetyl-CoA/CoA ratio has been postulated as the most important regulator of the enzymatic activity and specificity of HATs, rather than the absolute levels of acetyl-CoA
[50]. In brief, high intracellular acetyl-CoA levels would trigger histone acetylation, an epigenetic marker associated with open chromatin, activating oncogenes linked with BC progression, proliferation and migration
[20][30].
Another connection between metabolic processes and histone acetylation is provided by sirtuins (SIRTs), a type of NAD+-dependent histone deacetylases (HDACs)
[51]. The activity of these enzymes is closely linked with the NAD+/NADH ratio, and consequently with the energy status in the cell. For example, when glycolytic activity is enhanced, the NAD+/NADH ratio decreases, thereby inhibiting SIRT catalysis
[20][30]. The low NAD+/NADH ratio, together with an increase in HATs activity by elevated acetyl-CoA levels, could contribute to histone hyperacetylation and therefore an aberrant gene expression in BC
[30].
2.3. Role of Metabolites in the Nucleus
Finally, the translocation or production of commonly cytosolic metabolic effectors in the nucleus can supply essential intermediates to epigenetic machinery in specific chromatin regions, which affects gene expression. Increased SAM levels in the nucleus support epigenetic methyltransferase activity at specific regions of chromatin
[20]. This has been observed in cancer cells and is related to the translocation of splicing variants of MATs (S-adenosylmethionine synthetase, also known as methionine adenosyltransferase). Upregulated MAT1A levels have been reported in BC, specifically after treatment with chemotherapy, so MAT1A and possibly SAM could be related to the repression of tumor suppressor genes, (e.g., whose inhibition could confer tolerance or resistance to chemotherapy). Conversely, increased nuclear levels of acetyl-CoA can be produced by free diffusion of citrate or acetyl-CoA, but also by transient localization of the enzymes involved in its synthesis: ACSS2, ACLY, pyruvate dehydrogenase complex (PDC), and CAT (carnitine acetyltransferase). Post-translational modification of these enzymes within the nucleus or their association with lysine acetyltransferases (KATs) and transcription factors would explain their roles in chromatin regulation
[48]. When there is DNA damage, ACYL is phosphorylated within the nucleus, which promotes histone H4 acetylation near sites of DNA double-strand breaks to repair them. Therefore, in response to DNA damage, ACLY phosphorylation would be enhanced, which would allow an increase in the capture of citrate or acetyl-CoA in the nucleus
[48]. On the other hand, ACSS2 is recruited to specific genomic loci to supply acetyl-CoA for site-specific histone acetylation. Some studies have found that ACSS2 is translocated to the nucleus under low-glucose conditions upon phosphorylation by AMPK. Since cellular acetyl-CoA levels decrease when glucose is limited, a localized source of acetyl-CoA generated by ACSS2 could ensure the availability of this metabolite to KATs for histone acetylation
[48]. PDC acts as a co-activator of signal transducers and activators of transcription 5 (STAT5) proteins. STAT5 proteins regulate specific nuclear genes in response to growth factors and cytokines which are linked to crucial cellular functions such as proliferation, differentiation, and survival. The role of STAT proteins is underscored in the field of cancer because tumors have an aberrant constitutive activation of them, which significantly contributes to tumor cell survival and malignant progression of disease
[49]. Specifically in the context of BC, the findings obtained by Sun Y et al. suggested that the inhibition of STAT signaling by diindolylmethane (DIM) could decrease the invasiveness of BC, since DIM induced apoptosis in radioresistant cell lines. Therefore, DIM plus radiotherapy could be useful in overcoming such resistance
[50]. Other studies performed in BC cell lines using inhibitors against STAT3/5 such as Stattic, Nifuroxazide and SH-4-54 also showed reduced survival and increased apoptosis. In a xenograft model, Static monotherapy had effects on tumors, but its combination with chemotherapy had additive effects. These findings highlight that inhibitors against STAT3/5 are promising as novel mono- and combination therapies in BC
[51].
On the other hand, the regulation of NAD+/NADH levels in the nucleus is guaranteed by the activity of glycolytic enzymes (e.g., LDHA and glyceraldehyde 3-phosphate dehydrogenase (GAPDH)) since mitochondrial and nuclear membranes are impermeable to these cofactors
[20]. Within the nucleus, NADH could be implicated in regulatory processes associated with histone acetylation, which in turn would influence transcriptional activity.
Last, the role of pyruvate kinase embryonic isozyme M2 (PKM2) in BC has previously been highlighted. Monomeric PKM2 translocates into the nucleus where it functions as a protein kinase that phosphorylates histones during gene transcription and chromatin remodeling
[52]. Additionally, PKM2 upregulates the expression of c-Myc and cyclin D1, promoting the Warburg effect and cell cycle progression, respectively. Therefore, the role of nuclear PKM2 has been described as crucial for tumorigenesis, angiogenesis, and metastasis, and this protein has been postulated as a target for treating human cancers, including BC
[53]. Numerous studies have correlated PKM2 overexpression with the development and metastasis of BC through promoting cell proliferation, migration and invasion via the mitogen-activated protein kinase (MAPK) signaling pathway
[54], but also with advanced BC chemoresistance to cisplatin
[55] or anticancer efficiency to pirarubicin
[56]. Consequently, PKM2 could be a potential molecular prognostic marker of BC
[57].
In brief, metabolic enzymes in the nucleus link metabolic flux to gene regulation, and allow nuclear membrane-impermeable metabolites to be used in epigenetic processes
[58]. This metabolism–epigenetics axis would facilitate the adaptation to a changing environment around bladder tumors, providing a potential novel therapeutic target. The role that metabolites can play in modulating epigenetic enzyme action and gene expression is depicted in .
 +1 point
+1 point