The insulin receptor (IR) is critically involved in maintaining glucose homeostasis. It undergoes proteolytic cleavage by proprotein convertases, which is an essential step for its activation. The importance of the insulin receptor in the liver is well established, but its role in pancreatic β cells is still controversial.
1. Introduction
The IR is a key regulator of glucose homeostasis. The binding of insulin to its receptor in metabolic organs such as liver, pancreas, muscle, brain, and adipose tissue leads to the activation of a signaling pathway mostly aimed at maintaining glucose homeostasis in humans and animals
[1].
The IR is synthesized as an inactive precursor protein (proIR) that needs post-translational modifications to be activated. First, it requires endoproteolytic cleavage at the tetrabasic sequence RKRR
752, yielding disulfide-linked α and β subunits
[2]. Patients carrying a mutation in this cleavage site present with severe insulin-resistant diabetes
[3][4]. The tetrabasic sequence is most likely removed afterward by carboxypeptidase D, a process that has been shown to be required for full activation of the insulin-like growth factor 1 receptor
[5]. The endoproteolytic cleavage step has been shown to be performed by the proprotein convertase (PC) family members
FURIN, PACE4, PC5/6 (isoforms A and B), and PC7 in overexpression experiments
[6]. However, FURIN was put forward as the physiological PC for processing of the proIR in the secretory pathway and PACE4 at the cell surface when FURIN activity is reduced
[6]. In colorectal cell lines and in mouse mammary gland tissue, genetic ablation of
Furin indeed blocked proIR processing, suggesting non-redundant cleavage by FURIN
[7][8]. However, the proIR was normally processed in liver of an inducible
Furin knockout mouse model, revealing tissue-specific redundancy
[9]. Cleavage of the proIR by PCs in pancreatic β cells has not been investigated.
Insulin signaling has been studied extensively in many tissues. In liver, it regulates glucose metabolism by suppressing hepatic glucose production and promoting insulin clearance and glycogen storage
[10][11]. Liver-specific IR knockout mice show severe glucose intolerance together with hyperinsulinemia, and subsequent hepatic dysfunction
[12]. The physiological consequences of reduced IR signaling in β cells remain controversial. Notably, studies in transgenic mice have suggested an important role of the IR in pancreatic β cells for the regulation of peripheral glucose homeostasis
[13][14][15]. However, these studies made use of a Cre driver line shown to have an off-target expression in the brain
[16], or were affected by the presence of the human growth hormone (hGH) minigene
[17][18][19]. Recent studies, using Cre drivers lacking these pitfalls, showed that in β cells of adult islets, the IR controls insulin release and β-cell physiology
[20][21].
2. IR Processing and Signaling Are Severely Affected in FurKO β Cells
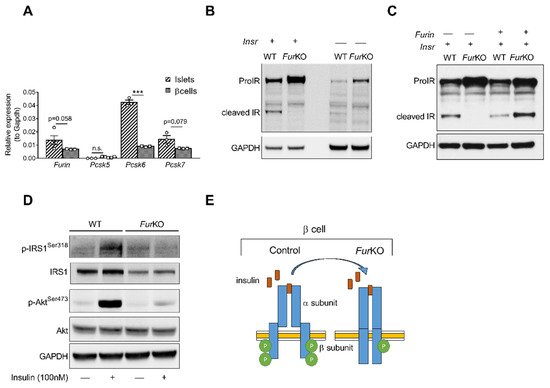
To determine the role of FURIN in the proteolytic activation of the proIR in pancreatic β cells, we first analyzed the murine β-cell line βTC3. This cell line showed a similar gene expression pattern of PCs active in the constitutive secretory pathway at mRNA levels compared to mouse islets (A). Furin, Pcsk6, and Pcsk7, encoding FURIN, PACE4, and PC7, respectively, were highly expressed, while Pcsk5, encoding PC5/6, was not. In βTC3 cells, the lack of Furin (FurKO) resulted in severely impaired, if not blocked, proIR cleavage (B), and transfection with recombinant Furin rescued the cleavage of mature IR (C). Importantly, the uncleaved proIR in FurKO β cells was unable to properly respond to insulin based on lack of phosphorylation of IRS1 and AKT after insulin stimulation (D,E).
Figure 1. IR processing and signaling are severely affected in FurKO β cells. (A) Comparative RT-qPCR analysis of Furin, Pcsk5, Pcsk6, and Pcsk7 gene expression in the islets of Furfl/fl mice (n = 3–5 per genotype) and wild-type βTC3 cells. *** p < 0.001 determined by two-way ANOVA with Sidak’s multiple comparisons test. All the RT-qPCR data were normalized to Gapdh. (B) Western blot analysis of the IR protein level in the whole-cell lysate of WT and FurKO IR-transfected and non-transfected βTC3 cells. GAPDH was used as loading control. (C) Western blot analyses of IR protein levels in whole-cell lysates from βTC3 cells transfected with Insr or co-transfected with Insr and Furin. GAPDH was used as loading control. (D) Western blot analysis of phosphorylated and total IRS1, and Akt in whole-cell lysates from βTC3 cells treated with 100 nM insulin or vehicle for 5 min. GAPDH was used as a loading control. (E) Schematic representation of a suggested mechanism of IR signaling in FurKO βTC3 cells compared to a physiological condition.
3. IR Proteolytic Cleavage Is Not Altered in Liver-Specific FurKO Mice
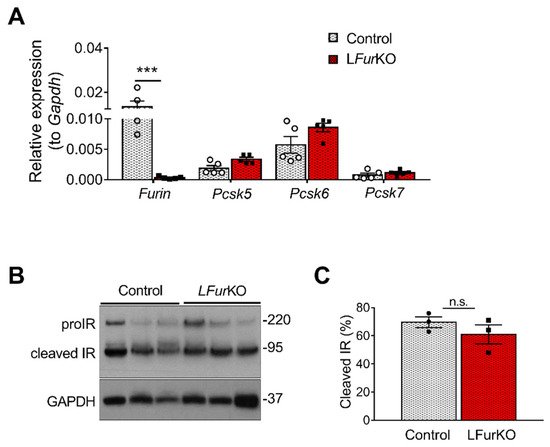
At mRNA level, Furin was the most abundant PC in mouse liver, although Pcsk5 and Pcsk6 were highly expressed as well (A). In vivo, conditional knockout of Furin in mouse hepatocytes using the Alb-Cre mice (LFurKO; A) induced a modest, non-significant increase of Pcsk5 and Pcsk6 gene expression (A). We subsequently evaluated the IR processing in hepatocytes from LFurKO and control mice by Western blot. As shown in B,C, the cleavage of proIR was not significantly reduced in LFurKO mice compared to the controls, indicating almost complete redundancy for proteolytic cleavage by other PCs.
Figure 2. IR proteolytic cleavage is not altered in the liver of Furin knockout mice. (A) RT-qPCR analysis of Furin, Pcsk5, Pcsk6, and Pcsk7 gene expression in the liver of either LFurKO or control (Furfl/fl) mice, n = 5 per genotype. *** p < 0.001 determined by two-way ANOVA with Sidak’s multiple comparisons test. All RT-qPCR data were normalized to Gapdh. (B) Western blot analysis of IR processing from liver membrane fractions of control and LFurKO mice, n = 3 animals per genotype. GAPDH was used as a loading control. (C) Protein-level quantification reported as % of cleaved IR over the total IR (ProIR + cleaved IR). ProIR data are represented as the mean ± SEM. Non-significant differences were detected by unpaired t-test.
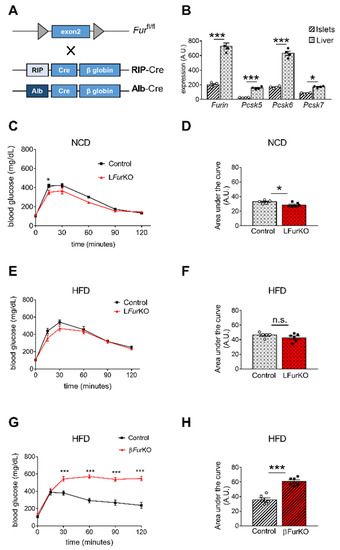
Figure 3. Impact of Furin knockout in liver and pancreatic β cells on glucose homeostasis (A) Schematic representation of the breeding approach. Mice in which the exon 2 of Furin had been floxed (Furfl/fl) were crossed with RIP-Cre or Alb-Cre driver lines. (B) Microarray analysis of Furin, Pcsk5, Pcsk6, and Pcsk7 gene expression in liver versus islets of control (Furfl/fl) mice, n = 4 per genotype. * p < 0.05; *** p < 0.001 determined by two-way ANOVA with Sidak’s multiple comparisons test. All the data were normalized to Gapdh. (C) Intraperitoneal glucose tolerance tests (IPGTTs) on 8-week-old male LFurKO and control mice on normal chow diet (NCD) (n = 5–6 mice/group). * p < 0.05 determined by two-way ANOVA with Sidak’s multiple comparisons test. (D) The area under the curve was expressed as g/dL/120 min; n = 5–6 mice/group. * p < 0.05 determined by unpaired t-test. (E) Intraperitoneal glucose tolerance test (IPGTT) on 16-week-old male LFurKO and control (Furfl/fl) mice fed for 8 weeks on a high-fat diet (HFD, 45% kJ from fat). No significant differences were found by two-way ANOVA with Sidak’s multiple comparisons test. (F) The area under the curve was expressed as g/dL/120 min; n = 5–6 mice/group. No significant differences were observed by unpaired t-test. (G) IPGTT on 16-week-old male βFurKO and control (Furfl/fl) mice fed for 8 weeks on a HFD (45% kJ from fat). *** p < 0.001 determined by two-way ANOVA with Sidak’s multiple comparisons test. (H) The area under the curve was expressed as g/dL/120 min; n = 6 mice/group. *** p < 0.001 determined by unpaired t-test. All data were presented as mean ± SEM.
4. Impact of Conditional Furin Deletion in Liver and Pancreatic β Cells on Glucose Homeostasis
β-cell-specific
Furin knockout (β
FurKO) mice were generated by breeding
Furfl/fl mice
[9] with RIP-Cre
+/− (A). A comparative gene expression analysis of all PCs active in the constitutive secretory pathway,
Furin,
Pcsk5,
Pcsk6, and
Pcsk7 in the liver and pancreatic islets of
Furfl/fl mice is displayed in B and . Overall, mRNA expression levels of all tested PCs were significantly higher in mouse liver with respect to isolated mouse islets. We subsequently studied how FURIN deficiency affected glucose homeostasis
in vivo in both tissue-specific knockout models. As expected, we observed that L
FurKO mice remained glucose tolerant both on chow and HFD (C–F) with normal insulin sensitivity on HFD (
Figure S1B). In contrast, β
FurKO mice were severely glucose-intolerant, with significantly higher fasting blood glucose levels on HFD (G–H and
Figure S1B), even on a chow diet, as we described in a previous study
[22]. We also did not observe changes in fasting blood glucose and body weight in L
FurKO mice compared to controls (
Figure S1C,D).
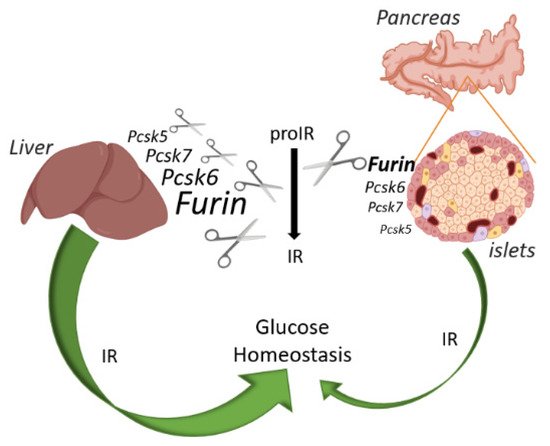
Figure 4. Schematic overview of the IR action on glucose and relative abundance of each PC in liver and pancreatic islets of mice. FURIN non-redundantly cleaves the proIR in pancreatic beta cells of mice, but the IR exerts only a minor effect on glucose homeostasis. In contrast, proprotein convertases other than FURIN can process proIR in the mouse liver, but here this receptor plays a pivotal role in glucose homeostasis. Image created with BioRender.com.
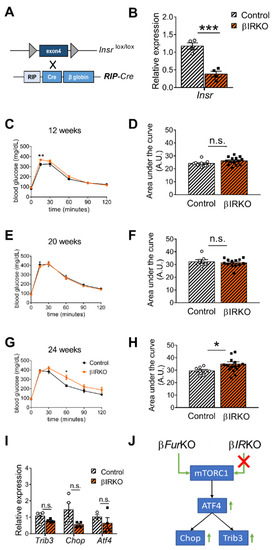
5. IR Deficiency in Pancreatic β Cells Does Not Induce Severe Glucose Intolerance
IR signaling has distinct roles in liver and pancreas
[12][14]. Since the role of the IR in pancreatic β cells is still controversial, we re-evaluated the effect of
Insr knockout in β cells of mice, using a Cre-driver line without the hGH minigene. We intercrossed RIP-Cre mice
[23] with
Insr-floxed (
Insrlox/lox) animals to generate βIRKO mice (A); these mice showed a 68% reduction of
Insr mRNA in the islets (B), consistent with (near) complete inactivation in the β cells. The remaining mRNA expression level was likely linked to the expression of
Insr in the other cell types of the islets (α, δ cells, and blood vessel endothelial cells), in which the Cre transgene is not expressed. In contrast to earlier observations
[13][24], glucose tolerance was not significantly altered in either 12- or 20-week-old mice (C–F), and only mildly affected in 24-week-old animals (G,H). In addition, fasting blood glucose levels and body weight were unaltered in β
IRKO mice (
Figure S1E,F). Moreover, a previous study reported that
Insr deficiency in mouse islets and β cell lines led to an induction of ATF4-dependent genes (i.e., Trib3)
[25], similar to our previous results using the β
FurKO
[22]. However, we observed a non-significant reduction in the gene expression levels of
Trib3,
Chop, and
Atf4 in isolated islets from the β
IRKO mice (I). This showed that the ATF4 pathway was not altered in these βIRKO mice (J).
Figure 5. Insr-knockout in pancreatic β cells (βIRKO) mildly impairs glucose tolerance in older mice. (A) Schematic representation of the breeding approach. Mice expressing the Insr gene in which the exon 4 had been floxed (Insrlox/lox) were crossed with the RIP-Cre driver line. (B) RT-qPCR analysis of Insr in the islets of control (Insrlox/lox) and βIRKO mice, n = 4 mice/group. All data were normalized to Gapdh and to the controls. *** p < 0.001 determined by unpaired t-test. (C–H) IPGTT on male βIRKO and control mice of 12 weeks (n = 9–14 mice/group) (C), 20 weeks (n = 7–13 mice/group) (E), and 24 weeks (n= 8–16 mice/group) (G). Quantification of the area under the curve for (C,E,G) was expressed as g/dL/120 min, and plotted in (D,F,H). * p < 0.05, ** p < 0.01 determined by two-way ANOVA with a Sidak’s multiple comparisons test.* p < 0.05 determined by unpaired t-test. All data were presented as mean ± SEM. (I) RT-qPCR analysis of Trib3, Chop, and Atf4 gene expression in the islets of control (Insrlox/lox) and βIRKO mice, n = 3–4 mice/group. No significant differences were detected by two-way ANOVA with Sidak’s multiple comparisons test. All the RT-qPCR data were normalized to the Gapdh. (J) Schematic of mTORC1/ATF4 pathway, which was upregulated in βFurKO but not in βIRKO mice.
 +1 point
+1 point