1000/1000
Hot
Most Recent
 +1 point
+1 point
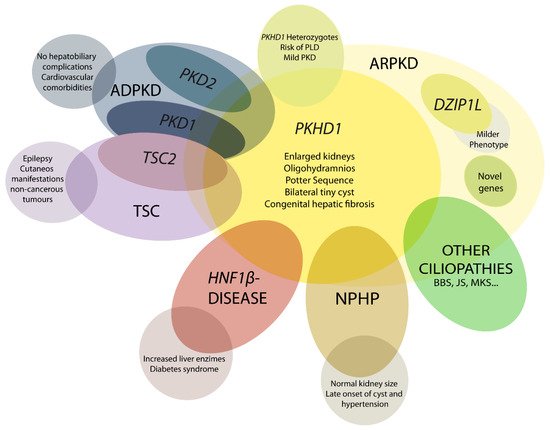
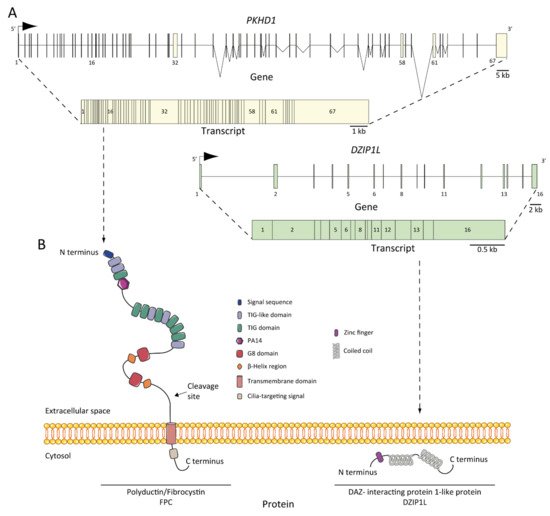
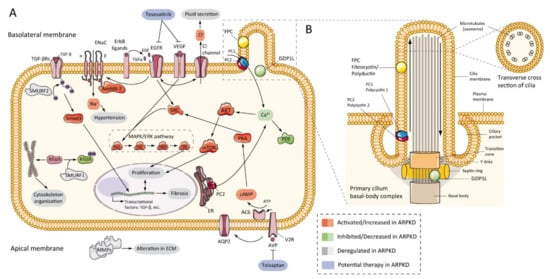
Autosomal recessive polycystic kidney disease (ARPKD) is a rare disorder and one of the most severe forms of polycystic kidney disease, leading to end-stage renal disease (ESRD) in childhood. PKHD1 is the gene that is responsible for the vast majority of ARPKD. However, some cases have been related to a new gene that was recently identified (DZIP1L gene), as well as several ciliary genes that can mimic a ARPKD-like phenotypic spectrum. In addition, a number of molecular pathways involved in the ARPKD pathogenesis and progression were elucidated using cellular and animal models. However, the function of the ARPKD proteins and the molecular mechanism of the disease currently remain incompletely understood.