Drug-induced liver injury (DILI) is an adverse reaction caused by exposure to drugs and herbal medicines or other xenobiotics. Depending on the presumed mechanism of action of the causative drug, DILI is typically classified as intrinsic (direct) or idiosyncratic [1], although indirect injury is emerging as a third type [2]. Intrinsic DILI is related to the cytotoxic properties of the causative drug or its metabolite(s). In this case, liver injury is dose-dependent and predictable, and damage can be reproduced in animal models [3]. Acetaminophen (APAP) toxicity is the most common cause for this type of DILI [4][5][6][7]. In contrast, idiosyncratic DILI is mostly host-dependent, multifactorial and unpredictable, since it is determined by both the properties of the drug and its interaction with environmental and host factors [8]. Idiosyncratic DILI is usually not dose-dependent, although the exposure to a threshold dose in each susceptible individual is necessary [9][10]. Moreover, the delay between starting the drug and the onset of clinical signs of liver injury is another characteristic of idiosyncratic DILI. Indirect liver injury is caused by an indirect action of the drug on liver or immune system, and can induce a new liver condition or exacerbate a preexisting one, such as worsening of hepatitis B or C.
The lines that distinguish types of hepatotoxicity are blurred, and majority of drug-induced liver reactions are considered idiosyncratic. Indeed, this is an unresolved issue and rather an academic classification as research over the last years has demonstrated that there are host susceptibility factors that influence the risk of intrinsic damage and, on the contrary, for drugs that are believed to cause idiosyncratic liver damage, there might be a dose threshold. Therefore, unless stated otherwise, the term DILI is used for idiosyncratic drug-induced hepatotoxicity in this review.
2. Potential Mechanisms Involved in DILI Pathogenesis
The liver plays an important role in metabolizing drugs or exogenous toxicants, protecting the organism from potential toxic chemicals [11]. Bioactivation processes of parent drugs rendering reactive metabolites and the mechanisms involved in detoxification and excretion of xenobiotics (most of them under genetic control) are critical for understanding the mechanisms of DILI [12]. However, many different hypotheses have been proposed due to the multivariant nature of the disease [13].
2.1. Drug Factors
Physicochemical and toxicological drug properties affect DILI risk
[14], contributing to initial cell damage that induces an adaptive and innate immune response. First, currently there is a consensus about the necessity of drug/metabolite exposure to a specific threshold level to DILI be initiated
[8]. In fact, there is an association between daily dose of a drug and poor DILI outcome
[10].
Drug lipophilicity is also associated with DILI risk, since it can enhance drug uptake from blood into hepatocytes, which results in an accumulation of reactive metabolites. In fact, a lipophilicity of LogP ≥ 3 in combination with high daily dose of drug (≥100 mg) are associated with severe DILI
[15].
Finally, the potential of a drug to form reactive metabolites is also associated with the pathogenesis of DILI
[16], due to their own toxicity and their ability to form drug-endogenous proteins adducts, which can activate the immune response
[17]. However, drugs unknown to form reactive metabolites, such as ambrisentan, flecainide, maraviroc, or bosentan can also cause DILI
[18].
2.2. Metabolic Mechanisms
Hepatocyte exposure to increased cellular stress is assumed to be the initial step in DILI development. Initial cell damage is induced by drugs and/or their reactive metabolites via covalent binding or direct damage to mitochondria, which leads to oxidative stress and the activation of stress-sensing signaling pathways, impairment of the mitochondrial function, and endoplasmic reticulum (ER) stress ().
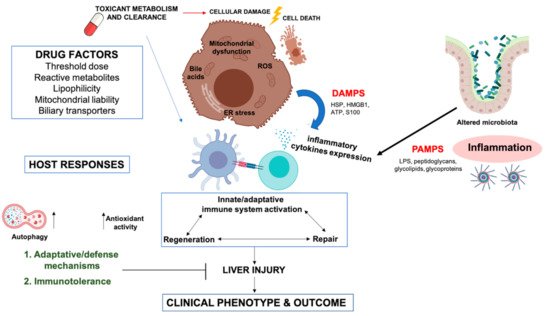
Figure 1. Cellular and molecular mechanisms involved in idiosyncratic drug-induced liver injury. Two key players in DILI, drug and host factors may interact in a multi-faceted manner at different functional pathways and determine individual susceptibility, clinical phenotype, and outcome. The hepatocyte damage caused by the action of drugs induce a complex, multivariant host response. First, cellular damage (oxidative stress, mitochondrial dysfunction, endoplasmic reticulum (ER) stress, and bile salt export pump (BSEP) inhibition, between others) can lead to cell death, provoking cell swelling and eventually rupture of the cell membrane, with the release of intracellular content, including damage-associated molecular patterns (damage-associated molecular patterns (DAMPs), such as high mobility group box protein 1 (HMGB1), heat shock proteins (HSP), ATP, S100 proteins, etc.) which stimulate a strong inflammatory/immune response. Inflammasome has also a very important role in development of liver injury, inducing cytokines secretion to attract and activate macrophages and neutrophils. Moreover, drugs can also alter intestinal microbiota (dysbiosis), and increase intestinal permeability, releasing bacterial products (Pathogen-associated molecular patterns, [PAMPs]) into the bloodstream. PAMPs (bacterial lipopolysaccharides (LPS), endotoxins, flagellin, etc.) act as costimulatory signals for the innate immune system activation. DAMPs and PAMPs are able to bind to TLR of the innate immune cells potentiating the immune response, cytokine release, and immune cell recruitment. Furthermore, drugs can form drug-endogenous proteins adducts that can act as neoantigens. Neoantigens presentation on specific HLA molecules could cause an adaptive immune response. Some HLA polymorphisms favor the presentation of drug-adducted neoantigens. Thus, individuals carrying the HLA variant are more susceptible to develop an adaptive immune response, typically leading to a T cell response directed at hepatocytes and usually involving cytotoxic CD8 T cells that target the peptide drug exposed on MHC class I molecules on the hepatocytes. Cellular damage also induces host adaptive and defense mechanisms, such as autophagy, antioxidant response and tissue repair. Moreover, because of its biological role with constant exposure to foreign antigens, the liver has a strong natural predisposition towards immune tolerance. This tolerance prevents a substantial immune response in the presence of the chemical insult, causing, at most, a mild liver injury that resolves spontaneously despite continued drug intake (i.e., adaptation). Clinically relevant liver injury is believed to result from a breakdown in hepatic immune tolerance. Concomitant inflammation can change the cytokine environment in favor of an immune response. Host factors such as age, gender, genetic factors, lifestyles, disease conditions, and co-medications are involved in the susceptibility of significant liver damage.
The mechanisms involved in the detoxification of drugs are critical in understanding the different processes triggered during DILI. The human CYP450 are membrane-bound proteins located in either the mitochondrial inner membrane or the smooth endoplasmic reticulum of hepatocytes, where they are responsible for the oxidation, peroxidation, and reduction being necessary for drug metabolism. The reactive metabolites generated during the metabolism of drugs are the main responsible for the sharp increase in oxidative stress directly generated in mitochondria of the injured hepatocyte
[19]. Increased reactive oxygen species (ROS) can directly damage DNA, proteins, enzymes, and lipids in cells and tissues and induce immune-mediated liver damage. Some drugs (e.g., valproic acid, [VPA]) can induce enhanced generation of ROS and triggering c-Jun N-terminal kinase (JNK). Thus, leading to hepatocyte death
[20]. This is a biphasic process: the early stage involves glycogen synthase kinase-3β (GSK-3β) activating mixed-lineage kinase-3 (MLK3), whilst the late phase is mediated by apoptosis signal-regulating kinase-1 (ASK1), thereby activating JNK
[21], which translocates to the mitochondria and triggers hepatocyte death, resulting in the amplification of mitochondrial ROS, such as in APAP-derived toxicity
[22].
Furthermore, damage-associated molecular patterns (DAMPs) released from injured hepatocytes activate innate immune responses, including cytokines such as tumor necrosis factor-alpha (TNFɑ), Fas ligand (FasL) or TNF-related apoptosis-inducing ligand (TRAIL)-expressing natural killer or natural killer T cells and neutrophils, which can activate death receptors such as TNF-R, FasR, and DR5. Besides, the activation of the necrotic cell death pathway is also present in DILI
[23]. Necrosis involves cell swelling, membrane bleb formation, and eventually the rupture of plasma membrane, causing the release of cellular components from necrotic cells that elicit an inflammatory response. Alternative mechanisms of regulated necrosis have emerged in recent years, such as necroptosis, pyroptosis, and ferroptosis. However, its relevance to acute or chronic DILI needs further research. Consequently, in DILI, altered cell functioning causes exacerbated ROS that further produce loss of hepatocyte function, and damaged hepatocytes release ROS, increasing overall oxidative stress, and ultimately leading to the activation of apoptotic and necrotic pathways
[23].
Metabolic activation of a drug and the generation of a reactive intermediate, the inadequate detoxification of the reactive intermediate, and the covalent binding to macromolecules can lead to subsequent liver toxicity
[24]. The formation of reactive metabolites has been well documented for drugs that have been withdrawn from the market or have a warning for hepatotoxicity. Examples are nefazodone, metabolized via CYP3A4 producing hydroxynefazodone, triazoledione, and m-chlorophenylpiperazine as metabolites or VPA, whose toxicity may be related to its metabolism through CYP2C9 and CYP2A6 to 4-ene-VPA
[24].
The metabolic activation of APAP is perhaps the best-documented case. Although APAP is metabolized to its glucuronidated and sulphated non-toxic metabolites in the liver, APAP overdose saturates these pathways, and the excess APAP is metabolized by CYP2E1 into the reactive metabolite N-acetyl-p-benzoquinoneimine (NAPQI), which is rapidly conjugated with glutathione (GSH), resulting in non-toxic mercapturic acid and cysteine conjugates that are excreted in the urine. When hepatic GSH levels are limited, free unconjugated NAPQI reacts with sulfhydryl groups on cysteine and lysine residues, generating NAPQI-protein adducts (APAP-protein adducts) in hepatocytes, particularly in mitochondria, leading to mitochondrial dysfunction
[25] and cell death.
Generally, the elevated level of chemically reactive intermediates overwhelms the capacity of the detoxification enzymes and endogenous antioxidants. A wide variety of antioxidant mechanisms to counterbalance pro- and antioxidant compounds, including GSH, superoxide dismutase (SOD), catalase, and glutathione peroxidase (GPX) that are pivotal in detoxification, are compromised, triggering imbalance between oxidants and antioxidants, and generating oxidative stress. Most importantly, mitochondria are the most affected hepatocyte organelles by GSH deficiency
[26].
2.3. BSEP Inhibition
The canalicular bile salt export pump (BSEP) is the only hepatocellular export system for primary bile salts into the canaliculus. BSEP inhibition has been proposed as a common mechanism of drug-induced cholestasis
[27][28], since a complete genetic deficiency of BSEP leads to cholestatic liver injury and liver failure
[29]. Evidence suggests that some drugs, such as bosentan
[30] or troglitazone
[31] induce DILI by inhibition of BSEP, supporting the hypothesis that the retention of bile acids in hepatocytes can induce cellular stress. On the other hand, the inhibition of BSEP or other bile salt transporters can initiate the immune response directly or through the release of DAMPs
[13]. However, BSEP inhibitory potency alone is not enough for determining DILI risk, and additional factors such as mitochondrial dysfunction
[32] or inhibition of multidrug resistance-associated proteins (MRP)
[33] should be considered.
2.4. Activation of the Immune Response
It has been increasingly clear that the immune response during DILI is determinant, since the presence of T cells and immune system activation in patients with DILI have been observed [14][34][35]. The immune response consists of a hypersensitivity reaction which provokes an inflammatory response that involves the innate and the adaptive immune system. Different hypotheses have been suggested to explain drug-induced immune system activation [23].
2.4.1. The Hapten Hypothesis
Haptens are small molecules that elicit an immune response only when covalent binding to endogenous proteins, forming adducts. Some drugs can form adducts by binding with endogenous proteins. When the drug-protein adduct (neoantigen) is taken up by antigen-presenting cells (APCs) and presented on major histocompatibility complex (MHC) class II proteins to T cells, it can elicit a subsequent adaptive immune response
[17]. However, this hypothesis alone cannot explain why only a minority of patients developed DILI induced by drugs described to form haptens. Moreover, flucloxacillin, a well-known hepatotoxicant, was found to have a hapten-dependent and a hapten-independent mechanism of adaptive immune activation, suggesting additional immunological pathways
[36].
2.4.2. The Danger Hypothesis
This hypothesis complements the hapten hypothesis and supports that, in order to precipitate an adaptive immune response, it is required an associated damage, a “danger signal”
[37]. This signal may include any intrahepatic or extrahepatic stressors, including ROS, mild inflammation or infection
[38].
After cell damage and cell death pathway activation, antigens derived from DAMPs are released, binding to their respective pattern-recognition receptors (PRRs) on the APCs. Depending on the endocytic pathway and the nature of the antigen, the APCs will present it to the T cells on MHC I or MHC II molecules stimulating the adaptive immune response
[39]. Besides, the danger hypothesis involves the costimulation of T cells by APCs through B7 receptors (CD80 and CD86) binding to CD28 on T cells. This costimulatory signal is required in order to induce an immune response instead of promoting immune tolerance
[40].
Additionally, DAMPs are able to bind to Toll-like receptors (TLR) of the innate immune cells potentiating the immune response, cytokine release, and immune cell recruitment
[41]. Among the main DAMPs found in the liver, the high mobility group box protein 1 (HMGB1), ATP, heat shock proteins (HSP), and S100 proteins are included
[42]. Binding to these receptors triggers downstream signaling pathways leading to the activation of caspase-1 and the consequent cleavage of proinflammatory cytokines including interleukins IL-1β and IL-18
[43], FasL, interferon gamma (IFNγ), and TNFɑ
[23].
2.4.3. The Pharmacological Interaction (p-i) Hypothesis
This hypothesis postulates that chemically inert drugs can activate certain T cells by specifically and directly forming non-covalent interactions with MHC molecules with which they fit with a sufficient affinity, triggering the activation of the immune system
[44][45][46][47]. In the last years, different drugs have been suggested to activate the immune system through p-i based stimulation
[36][44][48][49][50][51].
2.4.4. The Altered Peptide Repertoire Hypothesis
This hypothesis proposes that a drug can interact with MHC I molecules in a specific and non-covalent fashion and leads to presentation of altered endogenous peptides which elicit immune reactions. It can be regarded as a subset of p-i concept, but with the main key difference being the binding of novel self-peptides to the drug-MHC complex
[52].
2.4.5. The Multiple Determinant Hypothesis
This hypothesis suggests that DILI is dependent on the overlap of many different factors such as gender, age, genetics, environmental and physiological factors, etc. that increase the probability of an adverse hepatic event
[53][54]. Therefore, unless different factors concurred simultaneously, DILI will not develop. This might partially explain why the disease is so infrequent despite the risk genetic polymorphisms being common in the population.
2.4.6. The Inflammatory Stress Hypothesis
This hypothesis suggests that a potential inflammation occurring during drug treatment could interact with the action of the compound and produce liver injury [41]. Hepatic inflammation is often observed in DILI; therefore, it is suggested that DILI reactions could be unmasked by inflammation occurring during drug therapy. Inflammagens could bind to TLR or T-cell receptors (TCR), initiating the expression of inflammatory mediators. The inflammatory stress hypothesis has provided the first animal models in which liver injury is induced from different drugs associated with human DILI [55][56].
Moreover, a common mechanism of immune response involves activation of the inflammasome [57]. A recent study showed that the supernatant (presumably containing DAMPs) from the incubation of human hepatocytes with drugs that induce DILI activates the inflammasome in THP-1 cells, a macrophage cell line [58][59].
 +1 point
+1 point